Antonio Martín Parrilla DO, BSc (Hons) Osteopathy, BSc (Hons) Podiatry, BSc (Hons) Physiotherapy, MNeuSc, MSc.
DESCRIPCIÓN.
• Introducción: Los dolores de cabeza se diagnostican comúnmente basados en los signos y síntomas, cubriendo en realidad una amplia variedad de causas subyacentes. El ejemplo clásico sería el dolor de cabeza por tensión muscular, pero hay situaciones muy distintas tales como meningitis o accidente cerebrovascular que también debemos tener presentes. Esta sección se ocupa únicamente de la fisiopatología y la mejora de los dolores de cabeza benignos / primarios, los habituales (migraña, cefalea en racimos y otras cefaleas autonómicas trigeminales, cefaleas por componente alérgico, cefaleas de tipo tensional y otras cefaleas primarias no atribuidas a daño estructural); se hace hincapié en las migrañas como prototipo de estos trastornos. Una vez que se han excluido las causas patológicas graves del dolor de cabeza, el dolor de cabeza se puede tratar con medicamentos supresores de síntomas (que tienen riesgos y gastos asociados, sin beneficios colaterales) o mediante intervenciones biológicas / nutricionales / naturales que abordan los mecanismos causales subyacentes y así mejorar la salud en general.
• Importancia social y sanitaria: los dolores de cabeza y la migraña, aunque aparentemente insignificantes, en comparación con enfermedades potencialmente mortales como el cáncer y las enfermedades autoinmunes, representan enormes pérdidas en la calidad de vida y la productividad. El dolor de cabeza es un diagnóstico basado en la información subjetiva que nos aporta el paciente sobre su dolor de cabeza que se caracteriza por dos términos aparentemente simples: profundo o superficial. Las causas potenciales son numerosas, desde benignas tensiones musculares a potencialmente mortales, hemorragia intracraneal o meningitis. Nota importante: los pacientes con migraña tienen un mayor riesgo de enfermedades neurológicas y cardiovasculares (1,2), el tratamiento clásico del dolor migrañoso no aborda los trastornos bioquímicos, fisiológicos e inflamatorios subyacentes, mientras que las intervenciones nutricionales y antiinflamatorias tienen un gran potencial tanto para el alivio del dolor, como para la mejora de la salud general mediante la corrección de la fisiopatología subyacente.
PRESENTACIONES CLÍNICAS.
• Consideraciones generales sobre el dolor de cabeza: el dolor de cabeza, contempla una amplia gama de diagnósticos diferenciales, posibles causas y contribuciones; que van desde el simple «estrés«, la llamada «hipoglucemia reactiva«, hasta causas potencialmente mortales como apoplejía, aneurisma, tumores, meningitis… Especialmente en los dolores de cabeza de nueva aparición o de inicio agudo, las quejas subjetivas concomitantes (p. ej., letargo, somnolencia, cambios cognitivos / de humor, cambios en la visión) y / o presentaciones objetivas (p. ej., fiebre, erupción cutánea, galactorrea o déficits neurológicos) indican la necesidad de una evaluación adicional para excluir lesiones intracraneales importantes como adenoma hipofisario, meningitis, tumores o hematoma subdural. Dado que la vía sensorial del trigémino se activa en cualquier condición asociada con la inflamación del cerebro, el alivio del dolor de cabeza con analgésicos no excluye una enfermedad subyacente grave como hemorragia o meningitis.
• Migraña: se caracteriza por episodios recurrentes de dolor de cabeza de distribución unilateral, comúnmente con una sensación pulsátil; la gravedad varía de moderada a grave e invalidante; comúnmente comienzan en la adolescencia; asociado a herencia materna, debido a la herencia del ADN mitocondrial de la madre; dos veces más común en mujeres (5-25%) que en hombres (2-10%). La duración típica del «ataque» de migraña es de 4 a 72 horas. El 80% de la migraña es «migraña común» o «migraña sin aura«. La fotofobia y la fonofobia (sensibilidad excesiva a la luz y al sonido, respectivamente) son comunes, al igual que las náuseas, que a veces provocan vómitos. Los pacientes con migraña muestran un patrón constante de diferentes defectos mitocondriales aditivos y sinérgicos que afectan a varias ubicaciones en la vía de conversión del sustrato en energía celular / ATP: 1) deterioro enzimático de la citrato sintetasa (la primera enzima del ciclo de Krebs), 2) deterioro de la función de los complejos I-IV de la cadena de transporte de electrones, 3)deficiencia de coenzima Q-10 debido a una producción endógena insuficiente, lo que promueve el fallo en el desempeño de la cadena de transporte de electrones, así como una defensa antioxidante y antiinflamatoria reducida, 4)deficiencia de magnesio(que se observa en todos los tipos de dolor de cabeza), que conduce a un deterioro del complejo V(ATP sintasa) de la cadena de transporte de electrones, y también conduce a un aumento de la entrada de calcio intracelular después de la activación del NMDAr, lo que lleva a una mayor demanda metabólica en las neuronas.
• Migraña con aura: la migraña con aura se caracteriza por síntomas / déficits neurológicos focales; la localización y la afectación neurológica se ha atribuido tradicionalmente al vasoespasmo cerebral regional, que conduce a una reducción del flujo sanguíneo y al compromiso de la función neuronal / cerebral en las áreas afectadas. La migraña con aura puede presentarse con cualquier combinación: visión borrosa / alterada que incluye escotoma (punto ciego en la visión), la percepción de luces intermitentes u ondulantes, vértigo / mareos, alucinaciones como escuchar sonidos no reales o ver imágenes no reales. Parestesias, que por lo general se siente como un hormigueo en una mano o en un lado de la cara que se puede extender lentamente a lo largo de una extremidad. Dificultad del habla o del lenguaje, debilidad muscular. Algunos pacientes experimentan el aura como hiper / hipoactividad, depresión, antojos de comida, bostezos, cambios de humor. Más del 50% de los pacientes como resultado de la migraña refieren un deterioro significativo en las tareas de la vida y las relaciones personales.
• Cefalea en racimos: la cefalea en racimos (CR) o neuralgia de Horton, afecta predominantemente a hombres de mediana edad. Aunque la fisiopatología no está clara, según Current Medical Diagnosis and Treatment 2014, la activación de la sensación de dolor en el trigémino y la vasoconstricción están claramente involucradas, al igual que la migraña. Los pacientes con CR generalmente carecen de antecedentes familiares de dolor de cabeza o migraña. La CR se manifiesta con episodios de dolor periorbitario unilateral severo, generalmente con uno o más de los siguientes síntomas: congestión nasal ipsilateral, rinorrea (secreción nasal), lagrimeo, enrojecimiento del ojo y síndrome de Horner (ptosis palpebral / caída del párpado, miosis / constricción de la pupila y anhidrosis / reducción de la sudoración en el lado afectado). Los ataques de CR pueden ocurrir a diario (especialmente por la noche) durante varias semanas, y los pacientes a menudo se sienten inquietos y agitados. Los ataques de CR suelen durar de 15 minutos a 3 horas y ocurren en grupos durante semanas o meses y luego remiten sin motivo aparente. Los factores desencadenantes incluyen alcohol, estrés, deslumbramiento o alimentos específicos. El paciente característico con CR es un empresario masculino estresado, que fuma, con diferentes niveles de ingesta de alcohol… Mecánicamente, el estrés promueve la tensión muscular, especialmente en el cuello; el etanol / alcohol y el cianuro del humo del tabaco son toxinas mitocondriales; el empresario masculino estresado prototipo no come suficientes frutas y verduras para mantener la retención suficiente de magnesio. El hecho de que los pacientes con CR generalmente no tienen antecedentes familiares del trastorno, tienen un patrón de estilo de vida característico conocido por promover el deterioro mitocondrial y responden de manera rápida a la oxigenoterapia (obviamente una forma de apoyo mitocondrial, ya que la función principal del oxígeno en el cuerpo humano, es drenar protones de hidrógeno del espacio intramembranoso, mediante la formación de ATP y agua, durante la fosforilación oxidativa), estos pacientes tienen un deterioro mitocondrial secundario generado por el estilo de vida que conduce a sus dolores de cabeza, a través de la activación glial antes mencionada y la inflamación cerebral resultante y el resto de la cascada de eventos que induce el dolor. Todo esto apoya la afirmación de que: estos pacientes tienen un deterioro mitocondrial secundario generado por el estilo de vida, que conduce a sus dolores de cabeza, a través de la activación glial antes mencionada y la inflamación cerebral resultante y el resto de la cascada de eventos que induce el dolor.
DIAGNÓSTICOS DIFERENCIALES PRINCIPALES.
El diagnóstico diferencial de la cefalea mediante la anamnesis, el examen físico, las pruebas de laboratorio y por imágenes debe ser las claves diagnósticas. En particular, el examen neurológico, debe incluir una evaluación psicoemocional, así como un examen fundoscópico y de los pares craneales. Cualquier nuevo síntoma asociado al dolor de cabeza, incluso en un paciente con antecedentes de dolores de cabeza, debe recibir la debida diligencia por nuestra parte.
• Espondilosis cervical: la disfunción de la columna cervical y la artropatía pueden causar y contribuir al dolor de cabeza y cefaleas; consultar la historia clínica y el examen físico.
• Cefalea en racimos: se presenta con un intenso dolor periorbitario unilateral a menudo asociado con congestión nasal ipsilateral, rinorrea, lagrimeo, enrojecimiento de los ojos y síndrome de Horner transitorio / crónico; más común en hombres, especialmente en fumadores; exacerbado por el alcohol; tienden a repetirse a la misma hora.
• Dolor de cabeza por tos: dolor de cabeza severo y transitorio desencadenado por tos, esfuerzo, estornudos o risa. Los pacientes con molestias recurrentes deben ser evaluados con un examen neurológico completo y son candidatos para TAC / RM, ya que el 10% de los pacientes con dolor de cabeza persistente por tos tienen una lesión intracraneal (37).
• Trastornos dentales u oclusivos: valoración de la ATM, revisar la historia clínica, examen oral / dental.
• Depresión: buscar antecedentes que coincidan con la depresión: apatía, acontecimientos vitales estresantes recientes.
• Efectos secundarios medicamentosos: revisar cada medicamento para ver si los efectos secundarios se correlacionan con las quejas clínicas.
• Alergia alimentaria: evaluar con eliminación / provocación, revisar la historia clínica; considere análisis de sangre para casos recalcitrantes.
• Traumatismo craneoencefálico: evaluar con la historia clínica y el examen físico.
• Hiperparatiroidismo: comenzar por evaluar el calcio sérico.
• Hipertensión: evalúe la presión arterial; aunque la mayoría de los pacientes con hipertensión no tienen dolores de cabeza y la mayoría de los pacientes con dolores de cabeza no tienen hipertensión, aunque las exacerbaciones agudas de la hipertensión suelen precipitar el dolor de cabeza. Evaluar papiledema e hiperreflexia.
• Hipertiroidismo: pérdida de peso, temblores; evaluar TSH (generalmente baja) y T4 libre (siempre alta).
• Hipotiroidismo: evalúe TSH (típicamente elevado), T4 libre (generalmente bajo), T3 libre (puede ser bajo o normal), anticuerpos contra la peroxidasa tiroidea (TPO), se ve con hipotiroidismo autoinmune: enfermedad de Hashimoto; el tratamiento hormonal alivia la mayoría de los dolores de cabeza en pacientes con dolor de cabeza hipotiroideo.
• Infección por VIH: los pacientes con VIH tienen un mayor riesgo de contraer infecciones, incluidas infecciones intracraneales, en particular toxoplasmosis. El linfoma intracraneal también es más común en pacientes VIH positivos.
• Aneurisma intracraneal: puede presentarse con cefalea punzante; se evalúa mediante angiografía de contraste. En un gran estudio internacional con 1449 pacientes (39), el riesgo de rotura fue de menos del 1% por año, mientras que las complicaciones de la cirugía se observaron en aproximadamente el 14%. Un estudio japonés (40) encontró que el 95% de los pacientes tuvo un resultado favorable con la cirugía, lo que implica que el 5% tuvo un resultado desfavorable, que aún es mayor que el riesgo de ruptura (41). Un estudio más reciente también sugirió que los riesgos del tratamiento podrían exceder el riesgo de ruptura espontánea (42). Por lo tanto, el manejo clínico de los aneurismas intracraneales debe determinarse en base a cada paciente en particular, ubicación neuroanatómica, técnicas / tecnologías disponibles, investigación actual y experiencia del neurocirujano.
• Deficiencia de hierro: el hierro es necesario para el funcionamiento mitocondrial de la cadena de transporte de electrones, así como para la formación de los neurotransmisores dopamina y serotonina, de los cuales se puede decir que tienen un efecto analgésico. Como tal, la deficiencia de hierro puede promover dolores de cabeza en general y migrañas en particular; la deficiencia de hierro también podría contribuir a la presentación clínica de la fibromialgia (43). El estado óptimo de hierro se correlaciona con valores de ferritina sérica de 40-70 ng / ml. En raras ocasiones, hay personas que pueden presentar un defecto en el transporte de hierro de la barrera hematoencefálica al cerebro, en este caso necesitará tener un valor de ferritina sérica de 120 ng / ml para promover la entrada de hierro en el cerebro.
• Sobrecarga de hierro, con o sin hemocromatosis genética: resulta de especial interés someter a los pacientes a pruebas de sobrecarga de hierro. La sobrecarga de hierro provoca dolores de cabeza; la depleción de hierro puede aliviar los dolores de cabeza (45,46). El estado óptimo de hierro se correlaciona con valores de ferritina sérica de 40-70 ng / ml.
• Deficiencia de magnesio: la deficiencia de magnesio es común en los países industrializados (47,48,49,50) y se puede evaluar clínicamente (por ejemplo, dependiendo de la respuesta a la suplementación) o con pruebas de laboratorio (por ejemplo, magnesio intracelular). Los hallazgos asociados comunes con la deficiencia de magnesio son calambres musculares, bruxismo, estreñimiento, y antojos de dulces / caramelos y especialmente chocolate.
• Meningitis: evaluar mediante examen fundoscópico, hemograma completo y PCR; presentan erupción cutánea, fiebre. Si se sospecha meningitis resulta imperativo el traslado inmediato a urgencias.
• Migraña: la presentación clásica incluye periodicidad, unilateralidad, con pródromo, fotofobia, náuseas, vómitos, cambios visuales y antecedentes familiares positivos y aparición en la adolescencia temprana o la edad adulta; un gran porcentaje de pacientes con migraña no tiene la presentación clásica. La migraña puede asociarse con déficits neurológicos transitorios: entumecimiento, afasia, torpeza y debilidad.
• Tensión muscular y dolores de cabeza tensionales: evaluados con palpación / provocación de la musculatura cervical / craneal; empeoran con el estrés y al final de la jornada laboral; generalmente responde a terapias manuales, reducción del estrés, estiramiento de la musculatura afectada y suplementación con magnesio.
• Puntos gatillo miofasciales: Palpación / provocación de la musculatura cervical / craneal; tratar con estiramiento post-isométrico, mejorar la ergonomía postural, introducir Paleo dieta Mediterránea Vs dieta restrictiva empleando test TMH. Suplementación con vitamina D, calcio y magnesio.
• Trastornos oculares: evaluar antecedentes clínicos (p. ej., cambio reciente en la prescripción, gafas o lentes de contacto nuevas), examen neurológico, ocular y fundoscópico. Considerar la diabetes mellitus, la esclerosis múltiple o glaucoma. Es importante derivar de manera apropiada.
• Preeclampsia: el dolor de cabeza en una mujer embarazada puede indicar preeclampsia; evaluar la hipertensión, el edema y la proteinuria; en la mayoría de los casos, estará indicada la derivación obstétrica de emergencia.
• Feocromocitoma: la presentación común es cefalea periódica recurrente, con exacerbaciones por hipertensión, sudores y taquicardia / palpitaciones.
• Sinusitis o infección de los senos nasales: valorar los antecedentes, fiebre, dolor a la palpación de los senos nasales, secreción nasal; analítica completa y Proteína C Reactiva; considerar las imágenes radiográficas o de TAC.
• Arteritis temporal (AT), arteritis de células gigantes (ACG), polimialgia reumática (PMR): antecedentes de dolor difuso de cabeza / hombros y claudicación de la mandíbula, generalmente con síntomas sistémicos de mialgia y fatiga en un paciente mayor de 50 años; si se sospecha, debe evaluar la PCR / VSG y la palpación de la arteria. Hay que recordar que la arteritis temporal puede provocar ceguera; cualquier cambio visual en un paciente con AT / PMR debe considerarse una emergencia médica. «La pérdida de la visión es la manifestación más temida y ocurre con bastante frecuencia» (52).
• Disfunción de la articulación temporomandibular (ATM): valorar los antecedentes, examen oral / dental, dolor que empeora al masticar (diagnóstico diferencial con arteritis temporal); notablemente asociado con el exceso de glutamato intersticial.
• Otra lesión intracraneal del tumor: un tercio de los pacientes con tumores cerebrales presentan dolores de cabeza, por lo general empeoran al despertar y con esfuerzos. Evaluar con examen neurológico / imagenología.
MECANISMOS DEL DOLOR EN LA CEFALEA/MIGRAÑA.
Actualmente se informa que la vía común final para los dolores de cabeza «primarios» (p. ej., migraña y dolores de cabeza en racimos) están producidos por mediadores de inflamación neurogénica /cerebral: los mediadores de la inflamación del cerebro en general y los nervios periféricos, especialmente el trigémino (par craneal V), son generados a nivel neuronal, produciendo tanto la vasoconstricción como la sensación de dolor (3).
• El modelo contemporáneo integrador de la migraña: La tarea de los intelectuales es la creación de cohesión, integración y comprensión, por lo que una de las primeras tareas en la conversación sobre migraña es definir y caracterizar el trastorno. El tratamiento eficaz, salvo la suerte ciega, debe basarse en una comprensión integral y cohesiva del trastorno en su esencial totalidad. Los temas de los estudios experimentales y los ensayos clínicos deben integrarse / conciliarse, para que el mejor modelo fisiopatológico emerja triunfalmente por encima de las trivialidades de la anécdota y el dogma de la especulación farmacéutica. Más allá y además de la eficacia clínica, necesitamos una gran teoría unificada (GTU) que nos ayude a percibir la enfermedad y priorizar los tratamientos; de lo contrario, una comprensión desarticulada perpetuará el manejo médico desordenado, a la par que una dependencia farmacológica que ya observamos actualmente en el manejo de la migraña y el dolor de cabeza. Cada uno de los siguientes componentes está ordenado secuencialmente, de mayor a menor importancia: 1. disfunción mitocondrial, 2. Actividad glial sostenida, etc. Es importante destacar que, como señaló Thoreau en su conferencia publicada en 1849, Desobediencia Civil: «Amamos la elocuencia por sí misma, y no por ninguna verdad que pueda decir». No podemos quedarnos satisfechos con una explicación clara; la explicación debe tener un gran mérito en el mundo real, siendo probada por la seguridad y eficacia de los tratamientos que defiende. Este estándar revela la falsedad del modelo actual, tanto de la migraña como de la fibromialgia, ya que ambos modelos son selectivamente incompletos para justificar el tratamiento farmacológico y las intervenciones son innecesariamente peligrosas e inadecuadamente eficaces.
1. El deterioro mitocondrial es el origen de la migraña y la cefalea en racimos: los pacientes con migraña y cefalea en racimos, tienen defectos muy claros y constantes en el rendimiento mitocondrial, que conducen a una deficiencia de energía celular / ATP, producción excesiva de radicales libres “especies de oxígeno reactivo » (EOR), que promueven el daño celular y la inflamación). Los pacientes con migraña a menudo tienen deficiencia de coenzima Q-10 (CoQ-10), y esto causa disfunción mitocondrial y una protección antioxidante reducida contra los efectos dañinos y proinflamatorios de las EOR. Tratamientos nutricionales, como la riboflavina y CoQ-10, que apoyan la función mitocondrial, son consistentemente los tratamientos antimigrañosos más seguros y efectivos disponibles, lo que demuestra el origen mitocondrial de la migraña. Los defectos en la producción de energía celular/ATP hacen que las neuronas sean más inestables, lo que resulta en una activación excesiva, lo que ocasiona dolor y sensibilización.
La disfunción mitocondrial siempre promueve la inflamación, al menos aumentando la formación de radicales libres y la liberación de ATP libre a través de membranas mitocondriales con fugas; estas moléculas son percibidas por los receptores celulares como señales de peligro, lo que desencadena la respuesta de alarma inespecífica de la inflamación. Es probable que el deterioro metabólico contribuya a la vasodilatación, ya que las arterias se dilatan para aportar más oxígeno, para sustentar la demanda metabólica, en fisiología, esto se denomina “hiperemia reactiva”.
Durante los últimos años se está evidenciando, el modelo de activación microglial junto con la disfunción mitocondrial, los cuales están ganando fuerza; este modelo ayuda a explicar muchos aspectos divergentes de la migraña y proporciona unificación de modelos previamente fragmentados e inconexos. Uno de los principales impulsores de la migraña es la disfunción mitocondrial, que muestra una relación entre la gravedad y la respuesta, y es heredable por vía materna. La disfunción mitocondrial es suficiente para promover la activación microglial (células de Hortega: son células neurogliales del tejido nervioso con capacidad fagocitaria y de soporte). Las dos forman un círculo vicioso, promoviendo en última instancia la excitación neocortical y la sensibilización al dolor, promoviendo así nuevamente la continuación y el refuerzo de este círculo vicioso. Por analogía, los cerebros de estos pacientes son “fisiológicamente frágiles” con una constante inflamación; el cerebro está constantemente “ardiendo” (neuroinflamación crónica) o activamente «en llamas» (ataque migrañoso).
2. La activación glial sostenida es el resultado de la disfunción mitocondrial y causa inflamación e hiperexcitación del cerebro: las células gliales son el «pegamento«, las células interconectadas del cerebro compuestas principalmente por microglía y astrocitos. La microglía (las células inmunitarias del cerebro) son sensibles a las EOR y las señales inflamatorias, se «activan» (activación microglial) en respuesta a la inflamación periférica (incluida la obesidad, el trauma, la infección y la vacunación) y los eventos centrales «dentro del sistema nervioso» como trauma y estrés. Cuando la microglía se activa, envían señales a los astrocitos (células que apoyan física y químicamente a las neuronas) para que cambien el comportamiento, proporcionando menos apoyo protector a las neuronas del cerebro y provocando una mayor estimulación de estas mismas neuronas; más estimulación con menos protección hace que las neuronas se “sensibilicen”, eventualmente promueva el “agotamiento de estas neuronas”, ya que la sobreestimulación mantenida en el tiempo acaba produciendo “degeneración transneuronal” (es muy sencillo de entender, si utilizamos el ejemplo de los islotes de Langerhans, en el páncreas, al principio responden al exceso de insulina, aumentando su actividad, aparece el síndrome metabólico; pero esto continúa y se mantiene en el tiempo a lo largo de los años, haciéndolos trabajar por encima de sus ratios metabólicos, hasta que terminan por degenerar y morir, cuando ocurre la apoptosis celular, ya es demasiado tarde y es cuando aparece “la crónica de una muerte anunciada” la diabetes mellitus tipo 2; al principio no insulino dependiente y terminando por la insulino dependiente). La combinación de más excitación, más inflamación, menos energía / ATP y menos protección se denominan “excitotoxicidad” (lesión neuronal por sobreestimulación) y eventualmente conduce a neurodegeneración, daño a las neuronas, las estructuras cerebrales y el cerebro en su conjunto. Dicho de nuevo y de manera diferente: las células de la microglía en el cerebro reciben estímulos inflamatorios y luego activan los astrocitos para aumentar la estimulación de las neuronas a través del neurotransmisor excitador glutamato, que activa un receptor llamado receptor NMDA o lo que es lo mismo NMDAr (detallado más adelante); de esta manera, las señales inflamatorias se convierten en niveles alterados de neurotransmisores (especialmente glutamato, también ácido quinolínico (QUIN): un metabolito del triptófano producido durante condiciones de inflamación, reduce los niveles de serotonina, lo discutiremos más adelante) que estimulan a las neuronas a percibir más dolor. La estimulación excesiva de las neuronas se retroalimenta para causar más activación microglial, lo que resulta en un círculo vicioso.
3. La inflamación del cerebro y la inflamación neurogénica son el resultado de la activación glial y promueven una inflamación cerebral adicional: las células nerviosas se inflaman en respuesta a cualquier agresión; esto se llama neuroinflamación y promueve diversos trastornos neurológicos y psiquiátricos, como el dolor y la depresión (p. ej., los componentes del comportamiento de enfermedad), respectivamente. Las propias neuronas también pueden liberar mediadores inflamatorios; esto se llama inflamación neurogénica porque la inflamación proviene de las células nerviosas y al mismo tiempo afecta a esas mismas células nerviosas. Cuando se desencadena la inflamación del cerebro, afecta a todos los tipos de células principales del cerebro y se convierte en un ciclo de autorrefuerzo, a veces llamado “cerebro en llamas” (4). La inflamación en el cerebro tiene muchas consecuencias. Las neuronas inflamadas liberan neuropéptidos y mediadores inflamatorios que activan las células endoteliales (causando vasoconstricción) y promueven una inflamación adicional, la activación de mastocitos y plaquetas hace que estas células secreten aminas inflamatorias / vasoactivas, metabolitos del ácido araquidónico (como prostaglandinas, leucotrienos e isoprostanos); estas sustancias promueven una inflamación adicional y también promueven la constricción de los vasos sanguíneos. Sorprendentemente, la inflamación del cerebro y el deterioro metabólico que se observa en la migraña conocida como “depresión cortical propagada” (DCP) (fue inicialmente descrita por Leão en 1944, es una onda de excitación neuronal que se propaga a 2-6 mm/min en la corteza gris cerebral, esta despolarización neuronal explica el origen de los fenómenos corticales primarios que producen el aura), desencadena la liberación de la metaloproteinasa de la matriz enzimática (MME) asociada a la inflamación y que destruye los tejidos, que causa fugas en la barrera hematoencefálica (BHE), lo que provoca edema cerebral y una mayor absorción de moléculas inflamatorias de la sangre (5).
4. La disfunción mitocondrial y la activación glial se combinan para causar una alteración de la función de las neuronas cerebrales, desestabilización del cerebro y fragilidad metabólica; en su totalidad, esta combinación de tres partes de disfunción mitocondrial, inflamación glial y disfunción neuronal se denomina DCP: las neuronas son simultáneamente hiperactivas debido a la activación del receptor de neurotransmisores (NMDA) y también hiporreactivas debido al deterioro mitocondrial; esta “confusión fisiológica” contribuye a la función cerebral alterada que se observa en la migraña, especialmente la migraña con aura. En la migraña, el cerebro se “desestabiliza” (6,7), lo que lleva a lo que se puede llamar “fragilidad metabólica” o “fragilidad cerebral” que hace que los pacientes con migraña sean más sensibles a los cambios en la dieta, el clima, las hormonas, el estrés y el sueño. Estas combinaciones en el deterioro metabólico / mitocondrial con actividad cerebral aumentada / alterada (mediada principalmente por el glutamato sobre el receptor NMDA) e inflamación glial / neuronal / cerebral, es lo que crea la onda de función cerebral anormal: DCP, que tipifica la migraña y que promueve su exacerbación. La DCP conduce a la producción de enzimas inflamatorias y destructivas, como la MMP9 que causa fugas de la BHE y el subsecuente edema cerebral (secundario a la entrada de proteínas y agua en el cerebro), lo que a su vez origina una mayor entrada de moléculas que se encuentran en la sangre y que de otro modo no entrarían en el cerebro, este es el caso de las citoquinas proinflamatorias (8,9). El edema cerebral en la migraña está asociado a una reducción de la perfusión cerebral o reducción del flujo sanguíneo (10).
Muy importante, la neurotransmisión glutaminérgica aumentada es en sí misma suficiente para inducir DCP en modelos experimentales. Hay un artículo muy interesante publicado en 2014 que apoya este modelo (11), Malkov et al. demostraron que la DCP es causada por la producción de EOR, las cuales iniciaban el “colapso metabólico” en las células cerebrales.
5. La Ruta del Dolor: la envoltura del cerebro (meninges) es sensible a los cambios metabólicos e inflamatorios dentro del cerebro e interpreta las sustancias inflamatorias como señales de dolor: el nervio trigémino recibe transmisiones de las terminaciones nerviosas que rodean los vasos sanguíneos de la membrana que rodea el cerebro (piamadre) y dentro del cráneo (duramadre). Resulta importante recordar, como se mencionó anteriormente, que la barrera hematoencefálica se vuelve más permeable cuando el cerebro está inflamado, promoviendo así el paso / difusión de mediadores inflamatorios desde el cerebro a las neuronas cercanas que reciben estímulos nocivos y convierten la recepción de esas sustancias en impulsos nerviosos nociceptivos. Mientras que la inervación sensorial de la duramadre supratentorial se realiza a través de pequeñas ramas meníngeas del nervio trigémino, la inervación de la duramadre infratentorial se realiza a través de los nervios cervicales superiores (raíces C1, C2 y C3), lo que establece una relación entre el dolor de cuello, los estímulos intracraneales y la relación estomatognática. Es por ello que hablamos de la relación cráneo-mandibular y cráneo-cervical, en el Síndrome de Sensibilidad Central (SSC).
6. Sensibilización al dolor: a medida que se reciben más señales de dolor, el cerebro facilita la recepción de estos mensajes y, por lo tanto, se vuelve más sensible al dolor; esto se llama sensibilización central y se ve facilitado en gran medida por la inflamación del cerebro y el deterioro mitocondrial.
7. Dilatación y constricción de los vasos sanguíneos, y el efecto de los receptores de serotonina-1D: el deterioro metabólico puede desencadenar vasodilatación, mientras que los mediadores inflamatorios promueven la vasoconstricción; es por eso por lo que se ha observado vasodilatación y vasoconstricción en la migraña.
8. Los matices en la contribución de varios factores conducen a diferentes presentaciones clínicas; sin embargo, la disfunción mitocondrial e inflamación cerebral dominan la fisiopatología causal, por lo tanto, deben guiar el tratamiento: el modelo presentado y utilizado aquí es que la cefalea en racimos es simplemente una variante de la migraña. Con causas secundarias que amplían la primaria, la cual sigue siendo la disfunción mitocondrial subyacente. Estas causas secundarias son: una mayor contribución del estrés psicoemocional, la tensión muscular y las deficiencias nutricionales. La disfunción mitocondrial se observa tanto en la migraña como en la cefalea en racimos (12). Las deficiencias nutricionales (p. ej., Ácido fólico) y los excesos de inflamación sistémica y homocisteína sérica se observan en varios tipos de cefalea, tanto en población adulta como pediátrica (13).
MODELO FUNDAMENTAL DE LA MIGRAÑA.
1. Los pacientes con migraña muestran de manera muy clara y consistente evidencia de deterioro mitocondrial: esta disfunción mitocondrial genotípica puede deberse a diferentes factores, incluyendo 1) defectos en la síntesis de CoQ-10, 2) defectos en el ciclo del ácido cítrico, y 3) defectos en la función de la cadena de transporte de electrones… La mayoría de estos problemas pueden evitarse en parte o en gran medida mediante el uso de intervenciones nutricionales.
2. La disfunción mitocondrial promueve la inflamación en la microglia: La inflamación promovida por el exceso de radicales libres / especies de oxígeno reactivas (EOR) producidos por mitocondrias disfuncionales, promueve la activación microglial. La inflamación microglial causa disfunción mitocondrial a través del óxido nítrico (NO), causa deterioro de la enzima citocromo c oxidasa o complejo IV en la cadena de transporte de electrones…, que conduce a una reducción de la producción de energía celular / ATP y quizás directamente a través de citoquinas inflamatorias, creando así un círculo vicioso.
3. La activación de la microglía provoca la activación de los astrocitos, conduciendo a una liberación excesiva de glutamato.
4. El exceso de glutamato provoca una estimulación constante y mayor de las neuronas, promoviendo una descarga persistente y «recableado del cerebro», lo cual lo vuelve más sensible al dolor.
5. La hiperexcitación promueve el dolor, la depresión, fatiga, convulsiones, migraña y neurodegeneración. La combinación de disfunción mitocondrial con un exceso de excitación es particularmente devastadora para las neuronas y conduce a la muerte neuronal: neurodegeneración o también llamado degeneración transneuronal (DTN).
FISIOPATOLOGÍA: DEL PASADO AL MODELO ACTUAL.
La sensación de dolor de cabeza es el resultado de la activación y sensibilización de las neuronas sensoriales del dolor del trigémino que dan servicio a los vasos sanguíneos intracraneales y las meninges. Durante muchos años, el debate se centró en si predominaban las influencias vasculogénicas o neurogénicas; la mayoría, si no todos los dolores de cabeza parecen involucrar estos dos componentes principales, lo que permite el consenso de que los dolores de cabeza tienen un componente neurovascular. Dicho esto, el peso de la evidencia cambió cada vez más para respaldar el origen neurológico, desde el cerebro y las neuronas (en lugar de los vasos sanguíneos), de los dolores de cabeza en general y las migrañas en particular. Los «eventos iniciados por el cerebro«, como la depresión cortical propagada, una ola de perturbación eléctrica y metabólica que recorre la superficie del cerebro, haciendo que el tejido cerebral sea fisiológicamente inestable y, por lo tanto, más frágil y vulnerable a diversas agresiones, culmina en la producción del dolor, induciendo sustancias nociceptivas, incluidos iones de hidrógeno y metabolitos del ac. araquidónico, que irritan las neuronas sensoriales trigeminovasculares que rodean los vasos piales (14,15).
Se ha observado dilatación y constricción de las arterias en la migraña. La dilatación de las arterias puede ser una respuesta compensatoria temprana al deterioro de la producción de energía celular / ATP, a medida que la disfunción mitocondrial progresa de leve a más grave, a medida que los ciclos viciosos exacerban un defecto primario siempre presente. La vasodilatación en respuesta a la producción deficiente de energía / ATP es bien conocida en fisiología como «hiperemia reactiva«. A medida que la función mitocondrial se deteriora antes y durante un ataque migrañoso, pasa de un problema metabólico a un problema inflamatorio y las consecuencias de la disfunción mitocondrial (p. ej., EOR, inflamación, fallo de la homeostasis del calcio), generan más disfunción de las neuronas cerebrales debido a una excitación excesiva, lo cual conduce a vasoconstricción, específicamente a través del aumento del calcio intracelular en los astrocitos y la formación defosfolipasaA2 desencadenada por inflamación de prostaglandinas vasoconstrictoras, específicamente prostaglandina E2 (PGE2) y F2-alfa (PGF2), que también se elaboran a partir de tejido endometrial, apoyando así la base bioquímica de la migraña menstrual (16).
La inflamación neurogénica (en este diálogo bioquímico, la liberación de neuropéptidos de las neuronas del nervio trigémino a los vasos sanguíneos locales y las meninges) también es importante y contribuye a un círculo vicioso de dolor e inflamación (17). Los niveles elevados de calcio intracelular, que desencadenan vías inflamatorias, pueden ser promovidos por ac. araquidónico, hiperparatiroidismo secundario debido a deficiencia de vitamina D, una insuficiencia relativa de magnesio y también por deterioro mitocondrial. La degranulación de los mastocitos libera mediadores inflamatorios como serotonina, prostaglandina 1-2 e histamina, que inducen la inflamación local y la activación de los nociceptores meníngeos (18,19) y podrían servir como un vínculo fisiopatológico entre el estrés emocional o la exposición a alérgenos y el dolor de cabeza (es decir, el vínculo entre estresores ambientales y dolor de cabeza). Los mastocitos también pueden ser activados por neuropéptidos que se originan a partir de neuronas en el parénquima / tejido cerebral. Otra prueba del papel de la inflamación local en la migraña es el hallazgo de una mayor actividad del factor de transcripción nuclear kappa B (NF-kB) en la sangre yugular de pacientes con migraña durante el episodio (20). El NF-kB es un mediador importante de la inflamación a través de su capacidad para mejorar la transcripción de genes que codifican mediadores inflamatorios (21). Este modelo, coincide con lo observado a menudo entre factores externos y biopsicosociales tales como exposición a luces brillantes, hipoglucemia, estrés, ansiedad, exposición a alérgenos y fluctuaciones hormonales con el desencadenamiento de dolores de cabeza nuevos o recurrentes. Otra apreciación de la génesis intraneuronal de los dolores de cabeza, tales como las migrañas, agudiza nuestro enfoque en los eventos que ocurren dentro de la neurona, de este modo, vemos como las capacidades bioenergéticas mitocondriales se encuentran particularmente deterioradas, generando un aumento del calcio intraneuronal y la elaboración de mediadores inflamatorios derivados de ácidos grasos poliinsaturados omega-6 (n-6). Con las contribuciones mitocondriales y eicosanoides (moléculas que actúan como mediadores de la respuesta inflamatoria, cuyo precursor es el n-6) al dolor de cabeza, podemos intervenir con un modelo nutricional empleando suplementos de ácidos grasos para mejorar la función mitocondrial y modular la producción de eicosanoides, las dietas modernas usualmente tienen una proporción 10:1 de ácidos grasos omega-6 a omega-3. El hecho de no apreciar estos mecanismos fisiopatológicos subyacentes obliga a los pacientes a confiar en la supresión de síntomas farmacológicos, mientras los procesos subyacentes permanecen sin abordar.
El fracaso históricamente documentado de los tratamientos para la migraña ha surgido en gran parte del modelo fisiopatológico incompleto de la enfermedad en el que se basan esos tratamientos. Sin pura suerte, un tratamiento basado en un modelo erróneo o incompleto no tiene ninguna posibilidad de proporcionar un beneficio importante y mucho óptimo. Cualquier lista de tratamientos médicos para la migraña revela un catálogo de caos: fragmentos de modelos incompletos e inconsistentes y la terapia resultante, es decir, medicamentos que abordan una pequeña fracción del problema; de ahí la baja eficacia y el alto riesgo de efectos adversos, al tener que subir la dosis efectiva del fármaco cada vez más.
Importante para la perpetuación de cualquier enfermedad en curso, son los círculos viciosos que son iniciados y mantenido; los tratamientos se deben centran en romper estos círculos viciosos al no hacerlo permite que la enfermedad se reinicie y se perpetúe, incluso después de una eficacia terapéutica limitada, de un tratamiento incompleto. Podría introducir el concepto de terapias de «doble cadena» o «triple cadena«… que rompen simultáneamente múltiples ciclos viciosos, en contraste con tratamientos como los medicamentos que se enfocan solo en una molécula o en una vía, es decir, las «cadenas» son vías bioquímicas y fisiológicas; cuanto más podamos optimizar el número máximo de vías, mayores serán nuestras oportunidades para restaurar y disfrutar de una salud óptima.
Una revisión de 2015 que analiza la fibromialgia (FM) y el síndrome de dolor regional complejo (SDRC) se centró en la «neuroinflamación neurogénica«, cuya definición / concepto esencial es que la actividad neuronal en general y sus efectos inflamatorios en particular, pueden volverse autónomos y autoperpetuarse en el tiempo. La neuroinflamación podría iniciarse de un modo exógeno, por así decirlo, por estrés o por un trauma y luego convertirse en un círculo vicioso dentro del sistema nervioso, que promueve el dolor crónico y la neurodegeneración (22). La existencia de neuroinflamación neurogénica se vuelve probable dentro de sistemas metabólicos y fisiológicos disfuncionales, y predispuestos (es decir, «cebados«). Tales «factores cebadores» incluyen claramente una dieta proinflamatoria, deficiencias de nutrientes (especialmente de vitamina B6, magnesio, vitamina D y CoQ10), disfunción mitocondrial y disbiosis. Por lo tanto, el tratamiento de trastornos inflamatorios y dolorosos persistentes, incluidos, entre otros, migraña, dolores de cabeza recurrentes, fibromialgia y síndrome de dolor regional complejo, debe centrarse en el tratamiento de estos factores que perpetúan la enfermedad.
• La importancia del glutamato y el receptor NMDA en los síndromes de dolor de cabeza, migraña y dolor crónico: el neurotransmisor excitador glutamato estimula las neuronas al unirse al receptor NMDA (N-metil-D-aspartato) (NMDAr). El glutamato excitador (que promueve el dolor, convulsiones, migraña, ansiedad y depresión) se puede convertir en inhibidor GABA (ácido gamma-aminobutírico, que tiene un efecto inhibidor y relajante sobre las neuronas y el cerebro en general), a través del enzima ácido glutámico descarboxilasa, que depende de la vitamina piridoxina (vitamina B6). La estimulación del receptor NMDA por glutamato y otros como el ácido quinolínico (QUIN: es un «metabolito disfuncional» del aminoácido L-triptófano que se forma en respuesta a la inflamación y que causa inflamación adicional, daño oxidativo y neurotoxicidad) genera la entrada de calcio en las neuronaslo cual desencadena su activación. Una cantidad moderada de NMDAr es necesaria para el aprendizaje y la formación de recuerdos: función neurológica normal y saludable; sin embargo, demasiada estimulación con NMDAr provoca una sobreestimulación (excitotoxicidad) de las neuronas promoviendo así dolor, depresión, ansiedad, migraña, convulsiones / epilepsia y neurodegeneración. El magnesio y el zinc bloquean parcialmente el canal de calcio NMDAr para reducir / modular la entrada de calcio en las neuronas; de esta manera, se podría pensar que el magnesio y el zinc «suavizan el efecto» de la activación de NMDAr. La seguridad y eficacia de la suplementación con piridoxina (vitamina B6) para reducir la estimulación excesiva del NMDAr por el glutamato está más que demostrada, por tanto, requiere su inclusión en el tratamiento de todos y cada uno de los trastornos de dolor crónico, especialmente migraña y fibromialgia. La piridoxina hace más que simplemente reducir los niveles de glutamato, ya que también ayuda a reducir los niveles de homocisteína. El glutamato y la homocisteína activan al NMDAr y el receptor metabotrópico de glutamato 2 (mGluR2) (23). Generalmente, los niveles más altos de homocisteína se correlacionan con fatiga / dolor en pacientes con fibromialgia y síndrome de fatiga crónica, y con mayor riesgo de enfermedad cardiovascular en pacientes con migraña (24).
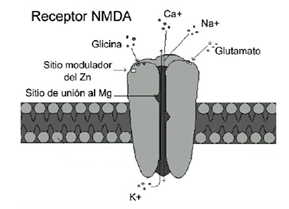
Fig 1. Receptor ionotrópico NMDA de glutamato, con cuatro subunidades proteicas y sitios distintos de unión para otros agonistas (p. ej., glicina, QUIN, homocisteína) y glutamato.
• En el tratamiento del dolor, incluidos los dolores de cabeza y la fibromialgia, la reducción de los efectos de la neurotransmisión mediada por el glutamato y sus efectos celulares es de gran importancia: el glutamato es un aminoácido con muchas funciones, incluida la función de precursor del antioxidante glutatión (GSH); también actúa como precursor del alfa-cetoglutarato (un sustrato para la producción de energía en el ciclo de Krebs) y también actúa como un neurotransmisor excitador. Nuestra preocupación es el papel del glutamato como estimulador de la neurotransmisión en el sistema nervioso central y periférico; mientras que una mínima estimulación glutaminérgica es normal y necesaria, el exceso de neurotransmisión glutaminérgica promueve muy claramente ansiedad, depresión, dolor por fibromialgia, dolor miofascial, puntos gatillo miofasciales, migraña, dolores de cabeza, convulsiones
y epilepsia; en el caso extremo, el exceso de glutamato en el cerebro provoca una sobreexcitación de las neuronas que conduce a la muerte celular (neurodegeneración) y daño cerebral leve o masivo / agudo o crónico.
Debido a que la neurotransmisión glutaminérgica promueve el dolor / ansiedad / depresión / neurodegeneración, nuestros objetivos terapéuticos son:
1. Reducir los niveles de glutamato con vitamina B6 y de este modo evitar / tratar la activación microglial (es decir, «inflamación del cerebro»).
2.Reducir la entrada de calcio provocada por el glutamato con zinc y magnesio, también vitamina D.
3. Alcalinización, aumento del consumo alimentos basificadores, como frutas y verduras que contienen citrato, el cual se convierte en bicarbonato para promover la alcalinización, uno de cuyos efectos es promover la retención de magnesio, aliviando así los dolores.
4.Tomar ácidos grasos omega-3 como el aceite de pescado para equilibrar la relación omega-3 Vs omega-6, para reducir mediadores de la respuesta inflamatoria, facilitando la producción de prostaglandinas tipo 3.
5.Reducir los efectos de la activación del receptor de glutamato / NMDA contrarrestando con la activación del receptor de benzodiazepina / GABA promoviendo la conversión de glutamato en GABA y quizás también usando niacinamida y productos fitoterápicos que actúan como ligandos para el receptor GABA.
SENSIBILIZACIÓN CENTRAL: 4 COMPONENTES PRINCIPALES DEL DOLOR
1. Señales de dolor: defectos en la producción de energía celular / ATP hace que las neuronas sean más inestables, lo cual genera un estado de activación excesiva, como respuesta aparece el dolor y la sensibilización. La disfunción mitocondrial siempre promueve la inflamación, aumentando la formación de radicales libres, que son percibidos por los receptores celulares como señales de peligro. El deterioro metabólico probablemente contribuye a la vasodilatación, ya que las arterias se dilatan para traer más oxígeno y poder soportar la demanda metabólica (en fisiología, esto se denomina «hiperemia reactiva«).
2. Inflamación cerebral: la microglía y astrocitos transforman las señales inflamatorias en el cerebro, alterando los niveles de neurotransmisores que activan aún más las neuronas.
3. Disfunción mitocondrial: las células nerviosas se vuelven inflamatorias en respuesta a cualquier estímulo; se llama neuroinflamación, promoviendo varios trastornos neurológicos y psiquiátricos. Las neuronas por sí mismas también pueden liberar mediadores inflamatorios; esto se llama inflamación neurogénica porque la inflamación viene de las células nerviosas. Cuando se desencadena la inflamación cerebral, afecta a todos los tipos de células principales del cerebro y se convierte en un ciclo retroalimentado, llamado “cerebro en llamas”. Los neuropéptidos liberados activan las células endoteliales, mastocitos y plaquetas para luego aumentar los niveles extracelulares de aminas, metabolitos del ac. araquidónico, péptidos e iones; estas sustancias promueven inflamación adicional y también vaso constricción.
4. Radicales libres, especies de oxígeno reactivas (EOR): «Se ha demostrado que la acumulación de EOR… es capaz de desencadenar un abrupto colapso metabólico (CM) que reproduce la mayoría de las características de la DCP. Esto sugiere que el estrés oxidativo puede ser la causa principal de la depresión cortical propagada y no solo su consecuencia. En condiciones patológicas, el fallo al neutralizar EOR durante la DCP puede resultar en el CM y posterior ignición de DCP. De hecho, los resultados in vivo muestran que cuando las EOR inducidas por el estrés oxidativo son reprimidas por un antioxidante exógeno, la aparición de DCP se reduce considerablemente (11).
• Varios tipos de receptores de glutamato en el sistema nervioso central y la periferia comparten los temas comunes de promover dolor e inflamación:
Los receptores de glutamato se describen en dos categorías:
1. Receptores ionotrópicos de glutamato (iGluR) divididos en tres grupos: receptores AMPA, NMDA y kainato; intercambian iones como el sodio y el calcio al activarse y, por lo tanto, se puede considerar que participan principalmente en la propagación de los impulsos nerviosos.
2. Los receptores metabotrópicos de glutamato (mGluR) (también con varios subtipos, como mGluR5) conducen más a la activación de vías intracelulares con resultados dependientes del tipo celular pero generalmente consistentes con algún tipo de activación celular y / o inflamación.
• Recepción de glutamato, con el receptor NMDA (NMDAr) como receptor prototipo:
Existen muchos tipos y subtipos de receptores de glutamato, y el subtipo específico NMDAr es claramente el más discutido por su relevancia, tanto en los trastornos de dolor crónico como en las enfermedades neurodegenerativas. El receptor de NMDA es activado por glutamato, el QUIN, aspartato, homocisteína y otras sustancias que actúan como agonistas / activadores o coactivadores / coagonistas como la D-serina o la glicina. Existen diferentes formas del receptor de NMDA en los sistemas nerviosos central y periférico, cada una con características y sensibilidad ligeramente diferentes a los agonistas y de los coagonistas. La imagen que presentamos es una versión conceptualizada pero precisa (26) y clínicamente relevante. Aunque tradicionalmente hemos pensado que los receptores de glutamato y NMDA existen por separado, es decir, en diferentes tipos de células de los receptores de GABA / benzodiazepínicos, ese hecho sigue siendo cierto, aunque algunas células están claramente dominadas por un tipo de receptor sobre otras, mientras que también vemos que los receptores de glutamato / NMDA pueden coexistir con los receptores de GABA / benzodiazepínicos en la misma célula y que estos receptores son interactivos, no simplemente opuestos, y ocasionalmente se comportan / interactúan de maneras paradójicas y específicas con la edad (27,28).
• La homocisteína (HYC), es un intermediario tóxico del metabolismo de los aminoácidos, activa los receptores de glutamato, lo que promueve el dolor, el dolor de cabeza / migraña / convulsiones:
El glutamato es el neurotransmisor excitador prototípico, que activa una amplia gama de receptores de glutamato ionotrópicos (incluido el receptor de NMDA) y receptores de glutamato metabotrópicos (mGluR) que están presentes en los sistemas nerviosos central y periférico y todos los cuales generalmente están involucrados en el dolor, intensificándolo. Tenemos una claridad absoluta de que tanto los receptores NMDA como el mGluR5 son activados por la homocisteína con el influjo de calcio resultante, al igual que con la activación mediada por glutamato de estos mismos receptores, Abushik et al (29). Por lo tanto, la elevación del calcio intracelular (Ca++) por HCY en las neuronas están mediadas por los receptores NMDA y mGluR5, mientras que las neuronas del ganglio cervical superior (SGC) se activan a través del subtipo mGluR5. Los efectos neurotóxicos a largo plazo en las neuronas periféricas y centrales involucran a ambos tipos de receptores. Esto tiene una importancia clínica muy alta, porque obtenemos la percepción de que la reducción de los niveles de homocisteína reducirá la estimulación total de estos receptores de glutamato en el cerebro, la médula espinal y la periferia; lo cual sugiere una reducción del dolor en la migraña y de fatiga en la fibromialgia.
• El glutamato promueve el dolor y la inflamación; por lo tanto, reducir los niveles de glutamato o reducir los efectos de su recepción son objetivos terapéuticos importantes, especialmente en el tratamiento del dolor, ansiedad / depresión, migraña y convulsiones / epilepsia:
Los niveles de glutamato aumentan por la inflamación de la microglía y la posterior activación de los astrocitos (30); por lo tanto, reducir la inflamación en general y la «inflamación del cerebro» específicamente es un objetivo terapéutico importante. La reducción de la inflamación debe centrarse siempre en el desencadenante de la inflamación, más comúnmente microbiana, disbiosis gastrointestinal (31) y / o metabólica, exceso de azúcar y «comida basura / rápida» en la dieta (32), deficiencia de vitamina D, falta de fitonutrientes debido a una ingesta insuficiente de frutas y verduras, ácidos grasos omega-3 insuficientes del aceite de pescado. El glutamato excita las neuronas y promueve el dolor, la depresión, la migraña, las convulsiones y la neurodegeneración; el glutamato se convierte fácilmente mediante el enzima ácido glutámico descarboxilasa (GAD) en ácido gamma-aminobutírico (GABA) que tiene efectos opuestos a los del glutamato, mediante la vitamina B6.
• Modulación de la entrada / acumulación de calcio después de la activación del receptor NMDA:
Después de la activación de NMDAr, entra sodio (Na +) para propagar los impulsos nerviosos, entra calcio (Ca ++) para generar la señalización intracelular, este mecanismo aumenta la promoción del dolor y las vías inflamatorias. El calcio intracelular es un famoso «segundo mensajero» responsable de procesos fisiológicos como la liberación pancreática de insulina; sin embargo, el exceso de calcio intracelular desencadena la activación de vías que pueden promover el dolor, la migraña y la hipertensión, de ahí el uso de fármacos bloqueadores de los canales de calcio (BCC) para tratar la migraña y la hipertensión. La entrada de calcio después de la estimulación con glutamato del NMDAr se reduce o «modula» tanto por zinc como por magnesio, los cuales se «adhieren» al NMDAr para reducir la entrada de calcio. Se describe a menudo el magnesio como un «corcho» o «tapón» del NMDAr. También se puede pensar que el magnesio (Mg) compite por el espacio con el calcio o bloquea algunos de los efectos del calcio intracelular; como tal, el Mg reduce el efecto de la estimulación del receptor de glutamato. El exceso de calcio intracelular también desafía o acentúa la capacidad de las mitocondrias, mientras que el magnesio apoya la función mitocondrial. El calcio intracelular promueve la contracción muscular, importante en la hipertensión (debido a la constricción sistémica de las arterias / arteriolas) y los puntos gatillo miofasciales (MFTP), una causa importante y contribuyente al dolor en la migraña y la fibromialgia, mientras que el magnesio promueve la relajación muscular, la dilatación arterial y alivio del dolor. Por lo tanto, esperaríamos, y de hecho vemos clínicamente, que la suplementación con magnesio (típicamente 600 mg por día para adultos) proporciona muchos beneficios al contrarrestar los efectos adversos de la estimulación glutaminérgica y el exceso de calcio intracelular, al tiempo que apoya la función mitocondrial. Muchos de los factores que contribuyen al exceso intracelular de calcio, son la deficiencia de vitamina D, deficiencia de magnesio, desequilibrio ácido-base, exceso de ácido araquidónico omega-6 en relación con los ácidos grasos omega-3. Todo esto se trata fácilmente con suplementos de vitamina D, suplementos de magnesio, la promoción de la alcalinización sistémica y la suplementación con ácidos grasos omega-3, respectivamente. Gracias a estas implementaciones conseguimos proporcionar analgesia, control de la tensión arterial y otros beneficios clínicos (33).
• Estimulación del receptor GABA / benzodiazepínico:
El efecto del neurotransmisor GABA en el receptor GABA, el cual también es afín a benzodiacepinas y fármacos barbitúricos, son bien conocidos.
Los receptores GABA también son activados por la forma niacinamida de la vitamina B3, así como por el alcohol / etanol. Los fitoterápicos que han demostrado un beneficio clínico a través de, al menos en gran parte, su activación del receptor de GABA / benzodiazepinas incluyen: Matricaria recutita (manzanilla), Melissa officinalis (toronjil), Passiflora incarnata (pasiflora), Piper methysticum (kawa-kawa), Scutellaria lateriflora (escutelaria), Valeriana officinalis (valeriana), Withania somnifera (ashwagandha) (34).
• Conversión de glutamato en GABA, la importancia de la vitamina B6 en la neuroprotección y el alivio del dolor:
La conversión de glutamato en GABA a través del enzima ácido glutámico descarboxilasa requiere de vitamina B6 (piridoxina), y la velocidad / eficiencia de esta conversión es generalmente proporcional a la provisión de B6. Dar más vitamina B6 da como resultado niveles más bajos de glutamato y, por lo tanto, menos activación del receptor de glutamato, lo que proporciona beneficios contra el dolor, la depresión y las convulsiones.
Como tal, lo que es obvio es que la suplementación con vitamina B6 claramente tiene un papel esencial en el tratamiento del dolor, la depresión, la migraña y las convulsiones. Todos los pacientes afectados por estos trastornos deben recibir suplementos de vitamina B6 de alta potencia, al menos como ensayo terapéutico, si no como un componente predeterminado de la terapia. Muy importante, la vitamina B6 alivia el dolor y el exceso de actividad cerebral que se observa con la migraña y las convulsiones, además de servir como cofactor del ácido glutámico descarboxilasa. La vitamina B6 también proporciona beneficios analgésicos y antiinflamatorios en los tejidos / nervios periféricos, en la médula espinal, en las estructuras cerebrales profundas del cerebro, como el tálamo el cual transmite el dolor (tracto espinotalámico lateral), así como el córtex.
• La activación del receptor de glutamato, especialmente el receptor de NMDA, en el sistema nervioso da como resultado la estimulación / despolarización de neuronas y la promoción de nuevas conexiones, promoviendo la memoria / aprendizaje y el dolor:
La activación del receptor NMDA permite que el sodio (Na +) y el calcio (Ca ++) entren en la célula. La entrada de sodio promueve la despolarización de la membrana nerviosa para permitir la propagación del impulso nervioso, a veces llamado «activación» nerviosa. La entrada de calcio después de la activación del receptor de glutamato desencadena eventos intracelulares, algunos de los cuales son beneficiosos para procesos como el aprendizaje y la memoria, mientras que otros, especialmente si los niveles de calcio intracelular son demasiado altos durante demasiado tiempo, son dañinos y promueven respuestas inflamatorias y estrés mitocondrial. Si bien normalmente hemos pensado que los receptores de glutamato y el receptor NMDA clásico existen en el cerebro y la neocórtex, ahora compartimos la claridad de que los receptores NMDA existen en todo el sistema nervioso, incluida la médula espinal y nervios periféricos. La activación de NMDAr es importante para el aprendizaje y la memoria, pero también es importante tener en cuenta que la generación de actividad neuronal / cerebral excesiva, asociada al exceso de glutamato, que se observa en convulsiones / epilepsia, dolor crónico y sobreactivación de neuronas que conduce a daño neuronal / cerebral: neuroexcitación, neurodegeneración y muerte neuronal por degeneración transneuronal.
• La activación del receptor de glutamato en los tejidos periféricos (fuera del sistema nervioso) no se comprende bien, pero nuevamente se correlaciona principalmente con el dolor y la inflamación:
Más allá del receptor NMDA y más allá del sistema nervioso, hay otros tipos de receptores de glutamato activos en todo el cuerpo. Dado que la mayoría de los tejidos y células están inervados por el sistema nervioso, la distinción entre el efecto glutaminérgico a través del sistema nervioso y el efecto directo sobre las células es un poco desafiante; sin embargo, uno de los temas más importantes observados en la literatura científica y de investigación es que los niveles elevados de glutamato en la periferia se asocian clara y causalmente con un aumento de la sensación de dolor y, en menor medida, con cambios fisiológicos adversos: la activación de mGluR5 en la piel se asocia con trastornos cutáneos inflamatorios e irritantes; este mismo subtipo mGluR5 también participa en la hipersensibilidad al dolor en enfermedades inflamatorias (35). Se observan niveles elevados de glutamato en enfermedades malignas (cáncer) y parece inhibir la función inmunológica; el bloqueo del receptor ionotrópico iGluR reduce la invasión del cáncer. Las asociaciones con una señalización excesiva de glutamato generan dolor e inflamación; cuando se inyecta glutamato en los músculos, el resultado es una mayor intensidad del dolor y un agrandamiento del campo receptivo (mayor área de sensibilidad al dolor) (36). Por lo tanto, se espera que los tratamientos para reducir los niveles de glutamato (especialmente vitamina B6) y reducir el efecto de los aumentos de calcio intracelular provocados por glutamato (principalmente, magnesio y vitamina D) reduzcan el dolor y la inflamación provocados por glutamato.
CONCLUSIÓN
La conversión de glutamato en GABA requiere vitamina B6 y proporciona beneficios analgésicos y «calmantes» para el cerebro y los músculos. El glutamato neuroexcitador se convierte en GABA neuroinhibidor por la enzima ácido glutámico descarboxilasa, que como se ha establecido en la epilepsia sensible a piridoxina, muestra una respuesta muy clara en su capacidad para reducir los niveles de glutamato en respuesta a la suplementación de vitamina B6 en dosis altas. El magnesio y el zinc (y quizás el cobre) retardan el paso del calcio a través de este canal, mitigando así algunos de los efectos de la activación de NMDAr. Apagar el óxido nítrico (ON) (por ejemplo, con la forma hidroxocobalamina de la vitamina B12), que de otro modo desencadenaría la liberación de glutamato. Controlar la activación glial (por ejemplo, con nutrientes antiinflamatorios como la vitamina D3 y EPA y DHA del aceite de pescado) que de otra manera promueven la producción de glutamato y QUIN son consideraciones importantes. La glicina se considera generalmente un coactivador necesario del NMDAr; pero dado que la glicina es ubicua y en su mayoría invariable, no se considera de gran relevancia como diana terapéutica clínica.
BIBLIOGRAFÍA
1. Fereshtehnejad et al. Comorbidity profile in dementia with Lewy bodies versus Alzheimer’s disease. Alzheimers Res Ther. 2014 Oct 6;6(5-8):65.
2. Chuang et al. Migraine and risk of dementia. Neuroepidemiology. 2013;41:139-45.
3. Tierney ML. McPhee SJ, Papadakis MA (eds). Current Medical Diagnosis and Treatment 2006, 45th Edition. New York: Lange Medical Books, 2006, pages 31-33.
4. Cohen G. The brain on fire? Ann Neurol. 1994 Sep;36(3):333-4.
5. Moskowitz MA. Genes, proteases, cortical spreading depression and migraine: impact on pathophysiology and treatment. Funct Neurol. 2007 Jul-Sep,22(3):133-6.
6. Malkov et al. Reactive oxygen species initiate a metabolic collapse in hippocampal slices: potential trigger of cortical spreading depression. J Cereb Blood Flow Metab. 2014 Scp;34(9):1540-9.
7. Moskowitz MA Genes, proteases, cortical spreading depression and migraine: impact on pathophysiology and treatment. Funct Neurol. 2007 Jul-Sep;22(3):133-6.
9. Wilson CJ, Finch CE, Cohen HJ. Cytokines and cognition the case for a head to toe inflammatory paradigm. J Am Geriatr Soc. 2002 Dec;50(12):2041-56.
10. Kim et al. Recurrent steroid responsive cerebral vasogenic edema in status migrainosus and persistent aura. Cephalalgia 2015 Jul 35:728-34. Ver también: Bereczki et al. Cortical spreading edema in persistent visual migraine aura. Headache. 2008 Sep;48:1226-9.
11. Malkov et al. Reactive oxygen species initiate a metabolic collapse in hippocampal slices: cortical spreading depression. J Cereb Blood Flow Metab. 2014 Sep;34(9):1540-9.
12. Lodi et al. Quantitative analysis of skeletal muscle bioenergetics and proton efflux in migraine and cluster headache. J Neurol Sci. 1997 Feb 27;146(1):73-80.
13. Nelson et al. Headache and biomarkers predictive of vascular disease in a representative sample of US children. Arch Pediatr Adolesc Med. 2010 Apr;164(4):358-62.
14. Moskowitz MA. Pathophysiology of headache-past and present. Headache. 2007 Apr;47 Suppl 1:S 58-63.
15. Moskowitz MA. Genes, proteases, cortical spreading depression and migraine: impact on pathophysiology and treatment. Funct Neurol. 2007 Jul-Sep 22(3):133-6.
16. Shaik MM, Gan SH. Vitamin supplementation as possible prophylactic treatment against migraine with aura and menstrual migraine. Biomed Res Int. 2015;2015:469529.
17. Tierney ML. McPhee SJ, Papadakis MA (eds). Current Medical Diagnosis and Treatment 2006, 45 h Edition. New York: Lange Medical Books, 2006, pages 31-33.
18. Levy D, Burstein R. Kainz V, Jakubowski M, Strassman AM. Mast cell degranulation activates a pain pathway underlying migraine headache. Pain. 2007 Jul;130(1-2):16.
19. Zhang XC, et al. Sensitization and activation of intracranial meningeal nociceptors by mast cell mediators. J Pharmacol Exp Ther 2007 Aug;322(2):806-12.
20. Sarchielli et al. NF-kappaB activity and iNOS expression in monocytes from internal jugular blood of migraine without aura during attacks. Cephalalgia. 2006 Sep; 1071-9.
21. Tak PP, Firestein GS. NF-kappaB. a key role in inflammatory diseases. J Clin Invest. 2001 Jan;107(1):7-11.
22. Littlejohn G. Neurogenic neuroinflammation in fibromyalgia and complex regional pain syndrome. Nat Rev Rheumatol. 2015 Nov;11(11):639-48.
23. Abushik et al. NMDA and mGluR5 in calcium mobilization and neurotoxicity of homocysteine in trigeminal/cortical neurons and glial cells. J Neurochem. 2014 Apr;264-74.
24. Moschiano et al. Homocysteine plasma levels in patients with migraine with aura. Neurol Sci. 2008 May;29 Suppl 1:S173-5.
25. Vormann et al. Supplementation with alkaline minerals reduces symptoms in patients with chronic low back pain. J Trace Elem Med Biol. 2001;15(2-3):179-83.
26. Vyklicky et al. Structure, function, and pharmacology of NMDA receptor channels. Physiol Res. 2014;63 Suppl 1:S191-203.
27. Ben-Ari et al. GABAA, NMDA and AMPA receptors: a developmentally regulated ‘ménage à trois! Trends Neurosci. 1997 Nov;20(11):523-9.
28. Ben-Ari Y. Excitatory actions of GABA during development: the nature of the nurture. Nat Rev Neurosci. 2002 Sep;3(9):728-39.
29. Abushik et al. The role of NMDA and mGluR5 receptors in calcium mobilization and neurotoxicity of homocysteine in trigeminal and cortical neurons and glial cells. J
Neurochem. 2014 Apr;129(2):264-74.
30. Béchade C. Cantaut-Belarif Y, Bessis A. Microglial control of neuronal activity. Front Cell Neurosci. 2013 Mar 28;7:32.
31. Crinnion, W. Results of a decade of naturopathic treatment for environmental illnesses. J Naturopathic Med 1994; 17: 21-27; Crinnion WJ. Environmental medicine, part one: the human burden of environmental toxins and their common health effects. Altern Med Rev, 5(1), 52-63.
32. Aljada et al. Increase in intranuclear nuclear factor kappaB and decrease in inhibitor kappaB in mononuclear cells after a mixed meal: proinflammatory effect. Am J Clin Nutr. 2004 Apr:79(4):682-90. Ver también: Mohanty et al. Glucose challenge stimulates reactive oxygen species (ROS) generation by leucocytes. J Clin Endocrinol Metab. 2000 Aug:85(8):2970-3.
33. Vasquez A. Intracellular Hypercalcinosis: A Functional Nutritional Disorder with Implications Ranging from Myofascial Trigger Points to Affective Disorders, Hypertension, and Cancer. Naturopathy Digest, 2006.
34. Sarris et al. Plant-based medicines for anxiety disorders, part 2: a review of clinical studies with supporting preclinical evidence. CNS Drugs. 2013 Apr,27(4):301-19.
35. Julio-Pieper et al. Exciting times beyond the brain: metabotropic glutamate receptors in peripheral and non-neural tissues. Pharmacol Rev. 2011 Mar;63(1):35-58.
36. Wang et al. Spatial pain propagation over time following painful glutamate activation of latent myofascial trigger points in humans. J Pain. 2012 Jun;13(6):537-45.
37. Tierney ML. McPhee SJ, Papadakis MA (eds). Current Medical Diagnosis and Treatment 2002, 41st Edition. New York: Lange; 2002. Page 999-1005.
38. Moreau et al. Headache in hypothyroidism. Cephalalgia 1998 Dec:687-9.
39. International UIA Investigators. Unruptured intracranial aneurysms-risk of rupture and risks of surgical intervention. N Engl J Med 1998 Dec 10:339(24):1725-33.
40. Orz et al. Risks of surgery for patients with unruptured intracranial aneurysms. Surg Neurol 2000 Jan;53(1):21-7; discussion 27-9.
41. International UIA Investigators. Unruptured intracranial aneurysms-risk of rupture and risks of surgical intervention. N Engl J Med 1998 Dec 10:339(24):1725-33.
42. Wiebers et al. Unruptured intracranial aneurysms: natural history, clinical outcome, and risks of surgical and endovascular treatment. Lancet. 2003 Jul 12:362(9378): 103-10.
43. Ortancil et al. Association between serum ferritin level and fibromyalgia syndrome. Eur J Clin Nutr. 2010 Mar;64(3):308-12.
44. Craig C. All My Test Results are Normal: A Smart Guide to Testing for Chronic Fatigue Syndrome. BookBaby. 2014.
45. Hagen et al. High headache prevalence among women with hemochromatosis. Ann Neurol 2002;51(6):786-9.
46. Stovner et al. Hereditary haemochromatosis in two cousins with cluster headache. Cephalalgia 2002 May;22(4):317-9
47. Innerarity S. Hypomagnesemia in acute and chronic illness. Crit Care Nurs Q. 2000 Aug;23(2):1-19.
48. Frankel et al. Hypomagnesemia in trauma patients. World J Surg- 1999 Sep,23(9):966-9.
49. Fox et al. An investigation of hypomagnesemia among ambulatory urban African Americans. Fam Pract. 1999 Aug;48(8):636-9.
50. Schimatschek HF, Rempis R. Prevalence of hypomagnesemia in an unselected German population of 16,000 individuals. Magnes Res. 2001 Dec;14(4):283-90.
51. Wright JV. Why you need 83 times more of this essential, cancer-fighting nutrient than the’experts’ say you do. Nutrition and Healing, 2005, vol. 12, no 4.
52. Tierney ML. McPhee SJ, Papadakis MA (eds). Current Medical Diagnosis and Treatment 2002. 47st Edition. New York: Lange; 2002, page 999-1005.
53. Tierney ML. McPhee SJ, Papadakis MA (eds). Current Medical Diagnosis and Treatment 2002, 47st Edition. New York: Lange: 2002, page 999-1005.
54. Tierney ML. McPhee SJ, Papadakis MA (eds). Current Medical Diagnosis and Treatment 2002, 47st Edition. New York: Lange: 2002, page 999.
55. Tierney ML. McPhee SJ, Papadakis MA (eds). Current Medical Diagnosis and Treatment 2006, 45th Edition. New York: Lange Medical Books, 2006, pages 31-33.
56. Tierney ML. McPhee SJ, Papadakis MA (eds). Current Medical Diagnosis and Treatment 2006, 45th Edition. New York: Lange Medical Books, 2006, pages 31-33.
57. Davidoff RA. Trigger points and myofascial pain: toward understanding how they affect headaches. Cephalalgia. 1998 Sep;18(7):436-48.
58. Tierney ML. McPhee SJ, Papadakis MA (eds). Current Medical Diagnosis and Treatment 2006, 45th Edition. New York: Lange Medical Books, 2006, pages 31-33
59. Vasquez et al. Clinical Importance of Vitamin D: Paradigm Shift for All Healthcare Providers. Altern Ther Health Med 2004; 10:28-37.
60. Barton JC, Edwards CQ, Bertoli LF, Shroyer TW, Hudson SL. Iron overload in African Americans. Am J Med. 1995 Dec;99(6):616-23.
61. Wurapa RK, Gordeuk VR, Brittenham GM, Khiyami A, Schechter GP, Edwards CQ. Primary iron overload in African Americans. Am J Med. 1996;101(1):9-18.
62. Baer DM, et al. Hemochromatosis screening in asymptomatic ambulatory men 30 years of age and older. Am J Med. 1995 May;98(5):464-8.
63. Olynyk J, Hall P, Ahern M, Kwiatek, MackinnonM. Screening for hemochromatosis in a rheumatology clinic. Aust NZ J Med 1994; 24: 22-5.
64. Phelps G, Chapman 1, Hall P, Braund W, Mackinnon M. Prevalence of genetic haemochromatosis among diabetic patients. Lancet 1989; 2: 233-4.
65. Kaikov Y, et al. Primary hemochromatosis in children: report of three newly diagnosed cases and review of the pediatric literature. Pediatrics 1992; 90: 37-42.
66. Edwards CQ, Kushner JP. Screening for hemochromatosis. N Engl J Med 1993; 328: 1616-20.
67. Gushuset TP, Triest WE. Diagnosis and management of precirrhotic hemochromatosis. W Virginia Med I 1990; 86: 91-5.
68. Balan V, Baldus W, Fairbanks V, et al. Screening for hemochromatosis: a cost-effectiveness study based on 12, 258 patients. Gastroenterology 1994;107:453-9.
69. Hagen K, Stovner LJ, Asberg A, et al. High headache prevalence among women with hemochromatosis: the Nord-Trondelag health study. Ann Neurol 2002 Jun;51(6):78
70. Stovner LJ, Hagen K, Waage A, Bjerve KS. Hereditary haemochromatosis in two cousins with cluster headache. Cephalalgia 2002 May 22(4):317-9.
71. Isobe C, Terayama Y. A remarkable increase in total homocysteine concentrations in the CSF of migraine patients with aura. Headache. 2010 Nov;50(10):1561-9.
72. DeQowin RL. DeQowin and DeQowin’s Diagnostic Examination. Sixth Edition. New York, McGraw-Hill; 1994, page 900.
73. Silberstein SD, et al. Efficacy and safety of topiramate for the treatment of chronic migraine. Headache. 2007 Feb;47(2):170-80.
74. Keays AC, Neher JO & Safranek S. Is osteopathic manipulation effective for headaches?. Clinical Inquiries, 2008 (MU).
75. Tierney ML. McPhee SJ, Papadakis MA (eds). Current Medical Diagnosis and Treatment 2002. 41st Edition. New York: Lange; 2002. Page 999-1005.
76. Daulatzai MA. Non-celiac gluten sensitivity triggers gut dysbiosis, neuroinflammation, gut-brain axis dysfunction, and vulnerability for dementia. CNS Neurol Disord Drug Targets. 2015;14(1):110-31.
77. Finsterer J, Leutmezer F. Celiac disease with cerebral and peripheral nerve involvement mimicking multiple sclerosis. J Med Life. 2014 Sep 15:7(3):440-4.
78. Hadjivassiliou et al. Headache and CNS white matter abnormalities associated with gluten sensitivity. Neurology. 2001 Feb 13:56(3):385-8.
79. Brown et al. White matter lesions suggestive of amyotrophic lateral sclerosis attributed to celiac disease. AJNR Am J Neuroradiol. 2010 May 31(5):880-1.
80. Adams et al. Nutrition education in U.S. medical schools: latest update of a national survey. Acad Med. 2010 Sep,85(9):1537-42.
81. Egger J, Carter CM, Wilson J, Turner MW, Soothill JF. Is migraine food allergy? A double-blind controlled trial of oligoantigenic diet treatment. Lancet 1983 Oct :2:865-9.
82. Monro J, Brostoff J, Carini C, Zilkha K. Food allergy in migraine. Study of dietary exclusion and RAST. Lancet 1980 Jul 5;2(8184):1-4.
83. Monro J, Carini C, Brostoff J. Migraine is a food-allergic disease. Lancet 1984 Sep 29;2(8405):719-21.
84. Finn R, Cohen HN. «Food allergy»: Fact or Fiction? Lancer 1978 Feb 25;1(8061):426-8.
85. Grant EC. Food allergies and migraine. Lancet 1979 May 5:1(8123):966-9.
86. Denton C. The elimination/challenge diet. Minn Med. 2012 Dec:95(12):43-4.
87. Shimada et al. Differential effects of repetitive oral administration of monosodium glutamate on interstitial glutamate concentration and muscle pain sensitivity. Nutrition 2015 Feb;31(2):315-23
88. Zhang et al. A mechanism of sulfite neurotoxicity: direct inhibition of glutamate dehydrogenase. Biol Chem. 2004 Oct 8;279(41):43035-45.
89. Harel et al. Supplementation with omega-3 polyunsaturated fatty acids in the management of recurrent migraines in adolescents. J Adolesc Health 2002 Aug;31(2):154-61.
90. Smith RS. The cytokine theory of headache. Med Hypotheses 1992 Oct;39(2):168-74.
91. Wagner et al. Prophylactic treatment of migraine with gamma-linolenic and alpha-linolenic acids. Cephalalgia. 1997 Apr;17(2):127-30.
92. Vaddadi KS. The use of gamma-linolenic acid and linoleic acid to differentiate between temporal lobe epilepsy and schizophrenia. Prostaglandins Med. 1981 Apr;6(4):3.
93. Al-Khamees et al. Status epilepticus associated with borage oil ingestion. J Med Toxicol. 2011 Jun;7(2):154-7.
94. Innerarity S. Hypomagnesemia in acute and chronic illness. Crit Care Nurs Q. 2000 Aug;23(2):1- 19.
95. Frankel H, Haskell R, Lee SY, Miller D, Rotondo M, Schwab CW. Hypomagnesemia in trauma patients. World J Surg. 1999 Sep,2309):966-9.
96. Fox CH, Ramsoomair D, Maboney MC, et al. An investigation of hypomagnesemia among ambulatory urban African Americans. J Fam Pract. 1999 Aug;48(8):636-9.
97. Schimatschek HF, Rempis R. Prevalence of hypomagnesemia in an unselected German population of 16,000 individuals. Magnes Res. 2001 Dec;14(4):283-90.
98. Boska et al. Contrasts in cortical magnesium, phospholipid and energy metabolism between migraine syndromes. Neurology 2002 Apr 23,58(8):1227-33.
99. Mauskop A, Altura BT, Altura BM. Serum ionized magnesium levels and serum ionized calcium/ionized magnesium ratios in women with menstrual migraine. Headache 2002 Apr;42(4):242-8.
100. Marcus JC, Altura BT, Altura BM. Serum ionized magnesium in post-traumatic headaches. J Pediatr 2001 Sep;139(3):459-62.
101. Boska et al. Contrasts in cortical magnesium, phospholipid and energy metabolism between migraine syndromes. Neurology 2002 Apr 23,58(8):1227-33.
102. Mazzotta G, Sarchielli P, Alberti A. Gallai V. Intracellular Mg++ concentration and electromyographical ischemic test in juvenile headache. Cephalalgia 1999 Nov;19(9):802-9.
103. Mishima K, et al. Platelet ionized magnesium, cyclic AMP, and cyclic GMP levels in migraine and tension-type headache. Headache 1997 Oct;37(9):561-4.
104. Peikert et al. Prophylaxis of migraine with oral magnesium. Cephalalgia 1996 Jun;16(4):257-63.
105. Mauskop A, Altura BT, Cracco RQ. Altura BM. Intravenous magnesium sulfate rapidly alleviates headaches of various types. Headache 1996 Mar 36(3):154-60.
106. Wang et al. Oral magnesium oxide prophylaxis of frequent migrainous headache in children: a randomized, double-blind, placebo-controlled trial. Headache. 2003;43:601-610.
107. Shahrami et al. Comparison of therapeutic effects of magnesium sulfate vs. dexamethasone/metoclopramide on alleviating acute migraine headache. J Emerg Med. 2015 Jan;48(1):69-76.
108. Sebastian et al. Estimation of the net acid load of the diet of ancestral preagricultural Homo sapiens and their hominid ancestors. Am J Clin Nutr 2002:76:1308-16.
109. Sebastian et al. Improved mineral balance and skeletal metabolism in postmenopausal women treated with potassium bicarbonate. N Engl J Med. 1994;330(25):1776-81.
110. Tucker et al. Potassium, magnesium, and fruit and vegetable intakes are associated with greater bone mineral density in elderly men and women. Am J Clin Nutr. 1999;69(4):727-36.
111. Vormann J, Worlitschek M, Goedecke T,Silver B. Supplementation with alkaline minerals reduces symptoms in patients with chronic low back pain. J Trace Elem Med Biol. 2001;15(2-3):179-83.
112. Talebi M, Goldust M. Oral magnesium, migraine prophylaxis. J Pak Med Assoc. 2013 Feb;63(2):286.
113. Johnston CS, Martin LJ, Cai X. Antihistamine effect of supplemental ascorbic acid and neutrophil chemotaxis. J Am Coll Nutr. 1992 Apr;11(2):172-6.
114. Rosa AC, Fantozzi R. The role of histamine in neurogenic inflammation. Br J Pharmacol. 2013 Sep;170(1):38-45.
115. Hasanzadeh Kiabi et al. Can vitamin C be used as an adjuvant for managing postoperative pain? A short literature review. Korean J Pain. 2013 Apr;26(2):209-10.
116. Mohseni M. Use of vitamin C as placebo in anesthesiology. Anesth Pain Med. 2013 Winter 2(3):141.
117. Van der Kuy PH, et al. Hydroxocobalamin, a nitric oxide scavenger, in the prophylaxis of migraine: an open, pilot study. Cephalalgia.2002: 22:513-519.
118. Kuzminski et al. Effective treatment of cobalamin deficiency with oral cobalamin. Blood 1998 Aug 15:92(4):1191-8 bloodjournal.org/cgi/content/full/92/4/1191.
119. Gordon N. Cerebral folate deficiency. Dev Med Child Neurol. 2009 Mar:51(3):180-2.
120. Piyathilake et al. Indian women with higher serum concentrations of folate and vitamin B12 are significantly less likely to be infected with carcinogenic or high-risk (HR) types
of human papillomaviruses (HPVs). Int J Womens Health. 2010 Aug 9:2:7-12.
121. Ventura et al. N-Acetyl-cysteine reduces homocysteine plasma levels after single intravenous administration by increasing thiols urinary excretion. Pharmacol Res. 1999 Oct;40(4):345-50.
122. Morrell MJ. Folic Acid and Epilepsy. Epilepsy Curr. 2002 Mar;2(2):31-34.
123. Paknahad et al. Effects of Common Anti-epileptic Drugs on the Serum Levels of Homocysteine and Folic Acid. Int / Prev Med. 2012 Mar;3(Suppl 1):S186-90.
124. Lea et al. Effects of vitamin supplementation and MTHFR (C677T) genotype on homocysteine-lowering and migraine disability. Pharmacogenet Genomics. 2009 Jun:422-8.
125. Menon et al. Effects of dietary folate intake on migraine disability and frequency. Headache. 2015 Feb;55(2):301-9.
126. Zimmerman M, Bartoszyk GD, Bonke D, et al. Antinociceptive properties of pyridoxine. Neurophysiological and behavioral findings. Ann N Y Acad Sci. 1990;585:219-30.
127. Folkers et al. Enzymology of the response of the carpal tunnel syndrome to riboflavin and to combined riboflavin and pyridoxine. Proc Natl Acad Sci. 1984 Nov;81(22):7076-8.
128. Kuo MF, Wang HS. Pyridoxal phosphate-responsive epilepsy with resistance to pyridoxine. Pediatr Neurol. 2002 Feb;26(2):146-7.
129. Baxter P. Pyridoxine-dependent seizures: a clinical and biochemical conundrum. Biochim Biophys Acta. 2003 Apr 11;1647(1-2):36-41.
130. Lewis PJ. Pain in the hand and wrist. Pyridoxine supplements may help patients with carpal tunnel syndrome. BMJ. 1995 Jun 10;310(6993):1534.
131. Wang HS et al. Pyridoxal phosphate is better than pyridoxine for controlling idiopathic intractable epilepsy. Arch Dis Child. 2005 May;90(5):512-5.
132. Mills et al. Genotypic and phenotypic spectrum of pyridoxine-dependent epilepsy (ALDH7A1 deficiency). Brain. 2010 Jul;133(Pt7):2148-59.
133. Sadeghi et al. Effects of pyridoxine supplementation on severity, frequency and duration of migraine attacks in migraine patients with aura. Iran J Neurol. 2015 Apr 4;14:74-80.
134. Kurlemann et al. Disturbance of GABA metabolism in pyridoxine-dependent seizures. Neuropediatrics. 1992 Oct;23(5):257-9.
135. Dave et al. Pyridoxine deficiency in adult patients with status epilepticus. Epilepsy Behav. 2015 Nov;52(Pt A):154-8.
136. Kang-Yoon SA, Kirksey A. Relation of short-term pyridoxine HCI supplementation to plasma vitamin B-6 vitamers and amino acid concentrations in young women. Am J Clin Nutr. 1992 Apr;55(4):865-72.
137. Magaña-Villa et al. B-vitamin mixture improves the analgesic effect of diclofenac in patients with osteoarthritis: a double blind study. Drug Res. 2013 Jun;63(6):289-92.
138. Doll H, Brown S, Thurston A, Vessey M. Pyridoxine (vitamin B6) and the premenstrual syndrome: a randomized crossover trial. J R Coll Gen Pract. 1989 Sep 39(326):364-8.
139. Moore D, Wahl R. Levy P. Hypovitaminosis D presenting as diffuse myalgia in a 22-year-old woman: a case report. J Emerg Med. 2014 Jun;46(6):e155-8.
140. Von Känel et al. Vitamin D and central hypersensitivity in patients with chronic pain. Pain Med. 2014 Sep;15(9):1609-18.
141. Thys-Jacobs S. Alleviation of migraines with therapeutic vitamin D and calcium. Headache. 1994 Nov-Dec;34(10):590-2.
142. Thys-Jacobs S. Vitamin D and calcium in menstrual migraine. Headache. 1994 Oct;34(9):544-6.
143. Timms et al. Circulating MMP9, vitamin D and variation in the TIMP-1 response with VDR genotype. QIM. 2002 Dec;95(12):787-96.
144. Bui et al. Dr. Max’s Migraine Minimizer Program. 2019 Jan;23(9):675-7.
147. Leventis P, Kiely PD. The tolerability and biochemical effects of high‐dose bolus vitamin D2 and D3 supplementation in patients with vitamin D insufficiency. Scandinavian journal of rheumatology. 2009 Nov;38(2): 149-153.
146. Titus et al. 5-Hydroxytryptophan versus methysergide in prophylaxis of migraine. Eur Neurol.1986; 25:327-329.
147. Shaik MM, Gan SH. Vitamin supplementation as possible prophylactic treatment against migraine with aura and menstrual migraine. Biomed Res Int. 2015;2015:469529.
148. Ziaei S, Kazemnejad A, Sedighi A. The effect of vitamin E on the treatment of menstrual migraine. Med Sci Monit. 2009 Jan;15(1):CR16-9.
159. Gaul et al. Improvement of migraine symptoms with a proprietary supplement containing riboflavin, magnesium and Q10: a randomized, placebo-controlled, double-blind, multicenter trial. J Headache Pain. 2015 Dec;16:516.
150. Chayasirisobhon S. Use of a pine bark extract and antioxidant vitamin combination product as therapy for migraine in patients refractory to pharmacologic medication. Headache. 2006 May:46(5):788-93.
151. Gasbarrini et al. Beneficial effects of Helicobacter pylori eradication on migraine.
Hepatogastroenterology. 1998 May-Jun;45(21):765-70.
152. Tunca et al. Is Helicobacter pylori infection a risk factor for migraine? A case-control study. Acta Neurol Belg. 2004 Dec;104(4):161-4.
153. Pinessi et al. Chronic Helicobacter pylori infection and migraine: a case-control study. Headache. 2000 Nov-Dec;40(10):836-9.
154. Ciancarelli et al. Helicobacter pylori infection and migraine. Cephalalgia. 2002 Apr;22(3):222-5.
155. Sangiorgi et al. Abnormal platelet mitochondrial function in migraine with and without aura. Cephalalgia 1994 Feb; 21-3.
156. Pauling L. Orthomolecular psychiatry, Varying concentrations of substances normally present in human body may control mental disease. Science. 1968 Apr 19:160:265-71.
157. Ames et al. High-dose vitamin therapy stimulates variant enzymes with decreased coenzyme binding affinity (increased K(m)). Am J Clin Nutr. 2002 Apr;75(4):616-58.
158. Rozen et al. Open label trial of coenzyme Q10 as a migraine preventive. Cephalalgia 2002; 22(2):137-41.
159 Ye CQ, Folkers K, et al. A modified determination of coenzyme Q10 in human blood and CoQ10 blood levels in diverse patients with allergies. Biofactors. 1988 Dec;1:303-6.
160. Schoenen J. Jacquy J. Lenaerts M. Effectiveness of high-dose riboflavin in migraine prophylaxis. A randomized controlled trial. Neurology 1998;50(2):466-70.
161. Sherwood M, Goldman RD. Effectiveness of riboflavin in pediatric migraine prevention. Can Fam Physician. 2014 Mar;60(3):244-6.
162. Martinez-Esteve Melnikova et al. Riboflavin in cyclic vomiting syndrome: efficacy in three children. Eur / Pediatr. 2015 Jul 31. [Epub ahead of print]
163. Condò et al. Riboflavin prophylaxis in pediatric and adolescent migraine. J Headache Pain. 2009 Oct;10(5):361-5.
164. Kabbouche et al. Carnitine palmityltransferase II (CPT2) deficiency and migraine headache: two case reports. Headache. 2003 May;43(5):490-5.
165. Hagen et al. Acetyl-1-carnitine versus placebo for migraine prophylaxis: A randomized, triple-blind, crossover study. Cephalalgia. 2015 Oct;35(11):987-95.
166. Tarighat Esfanjani et al. The effects of magnesium, L-carnitine, and concurrent magnesium-L-carnitine supplementation in migraine prophylaxis. Biol Trace Elem Res. 2012 Dec;150(1-3):42-8.
167. Magis D. Ambrosini A, et al. A randomized double-blind placebo-controlled trial of thioctic acid in migraine prophylaxis. Headache. 2007 Jan;47(1):52-7.
168. León J, Acuña-Castroviejo D. Escames G, Tan DX, Reiter RJ. Melatonin mitigates mitochondrial malfunction. J Pineal Res. 2005 Jan;38(1):1-9.
169. Velling DA, Dodick DW, Muir JJ. Sustained-release niacin for prevention of migraine headache. Mayo Clin Proc. 2003 Jun;78(6):770-1.
170. Prousky J, Seely D. The treatment of migraines and tension-type headaches with intravenous and oral niacin (nicotinic acid). Nutr J. 2005 Jan 26;4:3.
171. Ozkurt et al. Efficacy of high-flow oxygen therapy in all types of headache: a prospective, randomized, placebo-controlled trial. Am J Emerg Med. 2012 Nov;30(9):1760-4.
172. Myers DE, Myers RA. A preliminary report on hyperbaric oxygen in the relief of migraine headache. Headache. 1995 Apr;35(4):197-9.
173. Wilson et al. Hyperbaric oxygen in the treatment of migraine with aura. Headache. 1998 Feb;38(2):112-5.
174. Eftedal et al. A randomized, double blind study of the prophylactic effect of hyperbaric oxygen therapy on migraine. Cephalalgia. 2004 Aug:639-44.
175. Manzoni GC. Cluster headache and lifestyle: remarks on a population of 374 male patients. Cephalalgia 1999 Mar;19(2):88-94.
176. Scharff L, Marcus DA, Masck BJ. A controlled study of minimal-contact thermal biofeedback treatment in children with migraine. J Pediatr Psychol.2002: 27:109-119.
177. Siniatchkin et al. Self-regulation of slow cortical potentials in children with migraine: an exploratory study. Appl Psychophysiol Biofeedback.2000; 25:13-32.
178. Sartory et al. A comparison of psychological and pharmacological treatment of pediatric migraine. BehavResTher1998;36:1155-1170.
179. Calandre et al. Trigger point evaluation in migraine patients: an indication of peripheral sensitization linked to migraine predisposition? Eur J Neurol. 2006 Mar;13(3):244-9.
180. Borg-Stein J. Cervical myofascial pain and headache. Curr Pain Headache Rep. 2002 Aug:6(4):324-30.
181. Davidoff RA. Trigger points and myofascial pain: toward understanding how they affect headaches. Cephalalgia. 1998 Sep;18(7):436-48.
182. Lewit K, Simons DG. Myofascial pain relief by post-isometric relaxation. Arch Phys Med Rehabil 1984 Aug;65(8):452-6. “Este es uno de esos artículos que hay que leer”.
183. Bronfort et al. Efficacy of spinal manipulation for chronic headache: a systematic review. I Manipulative Physiol Ther 2001 Sep;24(7):457-66.
184. Tuchin PJ, Pollard H, Bonello R. A randomized controlled trial of chiropractic spinal manipulative therapy for migraine. J Manipulative Physiol Ther 2000 Feb;23(2):91-5.
185. Bronfort et al. Efficacy of spinal manipulation for chronic headache: a systematic review. J Manipulative Physiol Ther 2001 Sep,24(7):457-66.
186. Tuchin et al. A randomized controlled trial of chiropractic spinal manipulative therapy for migraine. J Manipulative Physiol Ther. 2000 Feb;23(2):91-5.
187. Kirk CR, Lawrence DJ, Valvo NL. States Manual of Spinal, Pelvic and Extravertebral Technics. Second Edition. Lombard, Illinois: National College of Chiropractic: 1985.
188. Kimberly PE. Outline of Osteopathic Manipulative Procedures. The Kimberly Manual 2006. Kirksville College of Osteopathic Medicine. Walsworth Publishing.
189. Bergmann TF, Peterson DH Lawrence DJ. Chiropractic Technique. New York; Churchill Livingstone: 1993.
190. Gatterman MI. Chiropractic Management of Spine Related Disorders. Baltimore; Williams and Wilkins: 1990.
191. Endres et al. Acupuncture for tension-type headache. J Headache Pain. 2007 Oct; 8(5): 306-314.
192. Vickers et al. Acupuncture of chronic headache disorders in primary care: randomised controlled trial and economic analysis. Health Technol Assess. 2004 Nov;8(48): iii, 1-35.
193. Wonderling D, et al. Cost effectiveness analysis of a randomised trial of acupuncture for chronic headache in primary care. BMJ. 2004 Mar 27,328(7442):747.
194. Cabyoglu MT, Ergene N, Tan U. The mechanism of acupuncture and clinical applications. Int J Neurosci. 2006 Feb;116(2):115-25.
195. Streng et al. Effectiveness and tolerability of acupuncture compared with metoprolol in migraine prophylaxis. Headache. 2006 Nov-Dec;46(10):1492-502.
196. Randlov et al. Intensive dynamic training for females with chronic neck/shoulder pain. Clin Rehabil. 1998 Jun:200-10.
197. Abushik et al. The role of NMDA and mGluRs receptors in calcium mobilization and neurotoxicity of homocysteine in trigeminal and cortical neurons and glial cells. J
Neurochem. 2014 Apr;129(2):264-74.
198. Yang N et al. Novel Clinical Evidence of an Association between Homocysteine and Insulin Resistance in Patients with Hypothyroidism or Subclinical Hypothyroidism. PLoS One. 2015 May 4;10(5):e0125922.
199. Ventura et al. N-Acetyl-cysteine reduces homocysteine plasma levels after single intravenous administration by increasing thiols urinary excretion. Pharmacol Res. 1999 Oct;40(4):345-50.
200. Cady RK, Schreiber CP. Beach ME, Hart CC. Gelstat Migraine (sublingually administered feverfew and ginger compound) for acute treatment of migraine when administered during the mild pain phase. Med Sci Monit. 2005 Sep;11(9):P165-9.
201. Shrivastava R, Pechadre JC, John GW. Tanacetum parthenium and Salix alba (Mig-RL) combination in migraine prophylaxis. Clin Drug Investig. 2006;26(5):287-96.
202. Grossmann M, Schmidramsl H. An extract of Petasites hybridus is effective in the prophylaxis of migraine. Int J Clin Pharmacol Ther 2000 Sep,38(9):430-5.
203. Lipton et al. Petasites hybridus root (butterbur) is an effective preventive treatment for migraine. Neurology. 2004 Dec 28;63(12):2240-4.
204. Anderson et al. Toxicogenomics applied to cultures of human hepatocytes enabled an identification of novel petasites hybridus extracts for the treatment of migraine with
improved hepatobiliary safety. Toxicol Sci. 2009 Dec;112(2):507-20.
205. Utterback et al. Butterbur extract: prophylactic treatment for childhood migraines. Complement Ther Clin Pract. 2014 Feb;20(1):61-4.
206. Kiuchi et al. Inhibition of prostaglandin and leukotriene biosynthesis by gingerols and diarylheptanoids. Chem Pharm Bull (Tokyo) 1992 Feb;40(2):387-91.
207. Tjendraputra et al. Effect of ginger constituents and synthetic analogues on cyclooxygenase-2 enzyme in intact cells. Bioorg Chem 2001 Jun;29(3):156-63.
208. Srivastava KC, Mustafa T. Ginger (Zingiber officinale) in rheumatism and musculoskeletal disorders, Med Hypotheses. 1992 Dec;39(4):342-8.
209. Srivastava KC, Mustafa T. Ginger (Zingiber officinale) and rheumatic disorders, Med Hypotheses. 1989 May;29(1):25-8.
210. Mustafa T, Srivastava KC. Ginger (Zingiber officinale) in migraine headache. J Ethnopharmacol. 1990 Jul,29(3):267-73.
211. Hu et al. The Analgesic and Antineuroinflammatory Effect of Baicalein in Cancer-Induced Bone Pain. Evid Based Complement Alternat Med. 2015;2015:973524.
212. Liao et al. Benzodiazepine binding site interactive flavones from Scutellaria baicalensis root. Planta Med. 1998 Aug;64(6):571-2.
213. Sarris et al. Plant-based medicines for anxiety disorders, part 2: a review of clinical studies with supporting preclinical evidence. CNS Drugs. 2013 Apr;27(4):301-19.
214. Sicuteri et al. Beneficial effect of capsaicin application to the nasal mucosa in cluster headache. Clin J Pain. 1989;5(1):49-53.
215. Marks et al. A double-blind placebo-controlled trial of intranasal capsaicin for cluster headache. Cephalalgia. 1993 Apr;13(2):114-6.
216. Fusco et al. Repeated intranasal capsaicin applications to treat chronic migraine. Br J Anaesth. 2003 Jun;90(6):812.
217. Fusco et al. Preventative effect of repeated nasal applications of capsaicin in cluster headache. Pain. 1994 Dec;59(3):321-5.
218. Rigatelli et al. Primary patent foramen ovale closure to relieve severe migraine. Ann Intern Med. 2006 Mar 21:144(6):458-60.
219. Dubiel et al. Migraine Headache Relief after Percutaneous Transcatheter Closure of Interatrial Communications. J Interv Cardiol. 2007 Dec 18; [Epub ahead of print]
220. Leone et al. Melatonin versus placebo in the prophylaxis of cluster headache: a double-blind pilot study with parallel groups. Cephalalgia 1996 Nov;16(7):494-6.
221. Peres MF, Rozen TD. Melatonin in the preventive treatment of chronic cluster headache. Cephalalgia. 2001 Dec;21(10):993-5.
222. Vogler et al. Role of melatonin in the pathophysiology of migraine: implications for treatment. CNS Drugs. 2006;20(5):343-50.
223. Leone et al. Melatonin versus placebo in the prophylaxis of cluster headache: a double-blind pilot study with parallel groups. Cephalalgia 1996 Nov;16(7):494-6.
224. Peres MF, Rozen TD. Melatonin in the preventive treatment of chronic cluster headache. Cephalalgia. 2001 Dec:21(10):993-5.
225. Glaser et al. Testosterone pellet implants and migraine headaches: a pilot study. Maturitas. 2012 Apr;71(4):385-8.
226. Stillman MJ. Testosterone replacement therapy for treatment refractory cluster headache. Headache. 2006 Jun;46(6):925-33.
227. Craig C. All My Test Results are Normal: A Smart Guide to Testing for Chronic Fatigue Syndrome. BookBaby, 2014.
228. Schober et al. Blood mercury levels in US children and women of childbearing age, 1999-2000. JAMA 2003:289:1667-74.
229. Kristin et al. Chemical Trespass. Pesticide Action Network North America. Available at panna.org, See also: Body Burden: The Pollution in People. ewg.org/2006 Feb.
230. Crinnion WJ. Environmental medicine, part 1: the human burden of environmental toxins and their common health effects. Altern Med Rev. 2000 Feb;5(1):52-63.
231. Crinnion WJ. Environmental medicine, part 2: health effects of and protection from ubiquitous airborne solvent exposure. Altern Med Rev. 2000 Apr;5(2):133-43.
232. Crinnion WJ. Environmental medicine, part 3: long-term effects of chronic low-dose mercury exposure. Altern Med Rev. 2000 Jun;5(3):209-23.
233. Crinnion WJ. Environmental medicine, part 4: pesticides – biologically persistent and ubiquitous toxins. Altern Med Rev. 2000 Oct;5(5):432-47.
234. Elemental Mercury Vapor Poisoning — North Carolina, 1988, cdc.gov/mmwr/preview/mmwrhtml/00001499.htm.
235. Shih H. Gartner JC Jr. Weight loss, hypertension, weakness, and limb pain in an 11-year-old boy. J Pediatr. 2001 Apr;138(4):566-9.
236. Sterzl I, Prochazkova J. Hrda P, et al. Mercury and nickel allergy: risk factors in fatigue and autoimmunity. Neuro Endocrinol Lett. 1999;20:221-8.
237. Padlewska KK. Acrodynia. Last Updated: February 15, 2007 eMedicine emedicine.com/derm/topic592.htm Accessed October 25, 2007
238. Chugh KS, Singhal PC, Uberoi HS. Rhabdomyolysis and renal failure in acute mercuric chloride poisoning. Med J Aust. 1978 Jul 29:2(3):125-6.
239. Chiu VC, Mouring D, Haynes DH. Action of mercurials on the active and passive transport properties of sarcoplasmic reticulum. J Bioenerg Biomembr. 1983 Feb;15(1):13-2.
240. Shamoo AE, Maclennan DH, Elderfrawi ME. Differential effects of mercurial compounds on excitable tissues. Chem Biol Interact. 1976 Jan;12(1):41-52.
241. Kalra V, et al. Succimer in Symptomatic Lead Poisoning. Indian Pediatrics 2002; 39:580-585 indianpediatrics.net/june2002/june-580-585.htm.
242. Bradstreet et al. Case-control study of mercury burden in children with autistic spectrum disorders. J Am Physicians Surgeons 2003; 8: 76-79 jpands.org/vol8no3/geier.pdf.
243. Forman et al. A cluster of pediatric metallic mercury exposure cases treated with meso-2,3-dimercaptosuccinic acid (DMSA). Environ Health Perspect. 2000 Jun;108(6):575.
244. Miller AL. Dimercaptosuccinic acid (DMSA), a non-toxic, water-soluble treatment for heavy metal toxicity. Altern Med Rev. 1998 Jun;3(3):199-207.
245. Kotter I, Durk H, Saal JG, et al. Mercury exposure from dental amalgam fillings in the etiology of primary fibromyalgia. J Rheumatol. 1995:22:2194-5.
246. Cobbett CS. Phytochelatins and their roles in heavy metal detoxification. Plant Physiol. 2000;123:825-32 plantphysiol.org/content/123/3/825.
247. Merchant RE, et al Nutritional supplementation with Chlorella pyrenoidosa for patients with fibromyalgia syndrome: a pilot study. Phytother Res 2000 May:14:167-73.
248. Pore RS. Detoxification of chlordecone poisoned rats with chlorella and chlorella derived sporopollenin. Drug Chem Toxicol. 1984:7(1):57-71.
249. Morita K, Ogata M, Hasegawa T. Chlorophyll derived from Chlorella inhibits dioxin absorption from the gastrointestinal tract and accelerates dioxin excretion in rats. Enviror Health Perspect. 2001 Mar;109(3):289-94.
250. Morita K, Matsueda T. Iida T, Hasegawa T. Chlorella accelerates dioxin excretion in rats. J Nutr. 1999 Sep;129(9):1731-6.
251. Nakano S, Noguchi T, Takekoshi H, Suzuki G, Nakano M. Maternal-fetal distribution and transfer of dioxins in pregnant women in Japan, and attempts to reduce maternal
transfer with Chlorella (Chlorella pyrenoidosa) supplements. Chemosphere. 2005 Dec;61(9):1244-55.
252. Nakano et al. Chlorella (pyrenoidosa) supplementation decreases dioxin and increases immunoglobulin a concentrations in breast milk. J Med Food. 2007 Mar:134-42.